编辑丨coisini
蛋白质-多肽相互作用在细胞环境中无处不在,并在众多生物过程中发挥关键作用。
由于多肽具有免疫原性低、生产成本低等特点,在药物递送、医学影像、治疗制剂、靶向蛋白质降解及其他生物技术应用中常作为高效调节剂使用。
深入理解蛋白质-多肽相互作用机制,是开发多肽类治疗药物的基础。然而,这类相互作用具有弱结合性、瞬时性及环境依赖性等特点,使得蛋白质-多肽结合模式的高效虚拟筛选与结构表征面临巨大挑战。
最近,浙江大学侯廷军、康玉团队联合华盛顿大学、中南大学提出一种扩散生成模型 ——RAPiDock,在全原子水平上实现了理性、精准且快速的蛋白质-多肽分子对接。
研究论文以《Protein–peptide docking with a rational and accurate diffusion generative model》为题发表在《Nature Machine Intelligence》上。

论文地址:https://www.nature.com/articles/s42256-025-01077-9
RAPiDock 概览
RAPiDock 模型通过引入物理约束条件有效缩小了采样空间,并采用双尺度图(bi-scale graph)在保证效率的同时捕获多维结构信息。RAPiDock 还创新性地基于 Clebsch-Gordan 张量积构建网络架构,确保模型具备物理对称性。
如下图所示,RAPiDock 工作流通过输入肽段序列与蛋白质结构,可生成多肽结合构象的分布作为输出。
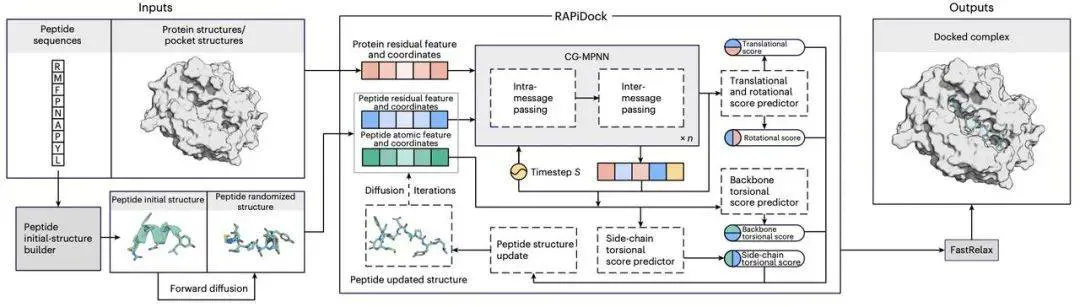
具体而言,RAPiDock 在对接涉及的自由度(包括肽段的全局平移、全局旋转以及主链 / 侧链扭转角)上定义了扩散过程。
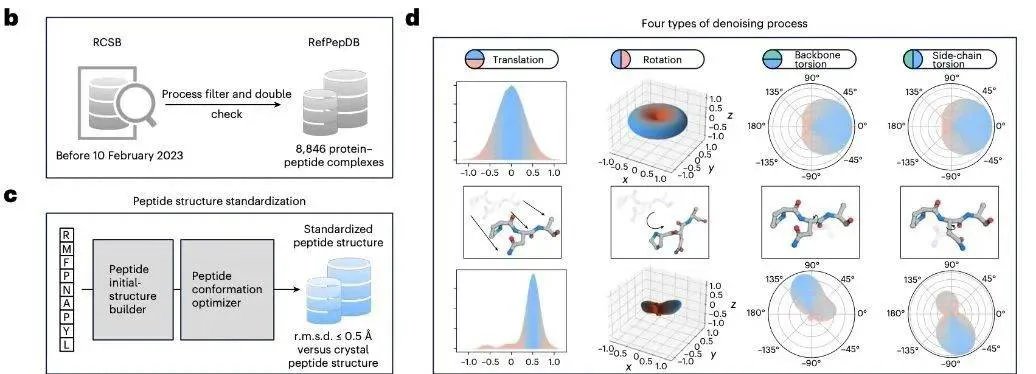
通过引入额外物理约束,RAPiDock 不仅有效缩小了采样空间,还提升了构象真实性。该研究采用双尺度图分别在残基和原子级别表征肽段,通过扩散过程将肽段构象从无信息的噪声先验分布逐步转化为有序的目标分布。
该研究还开发了肽段初始结构构建器,可将一维肽序列转化为标准化的三维初始结构。
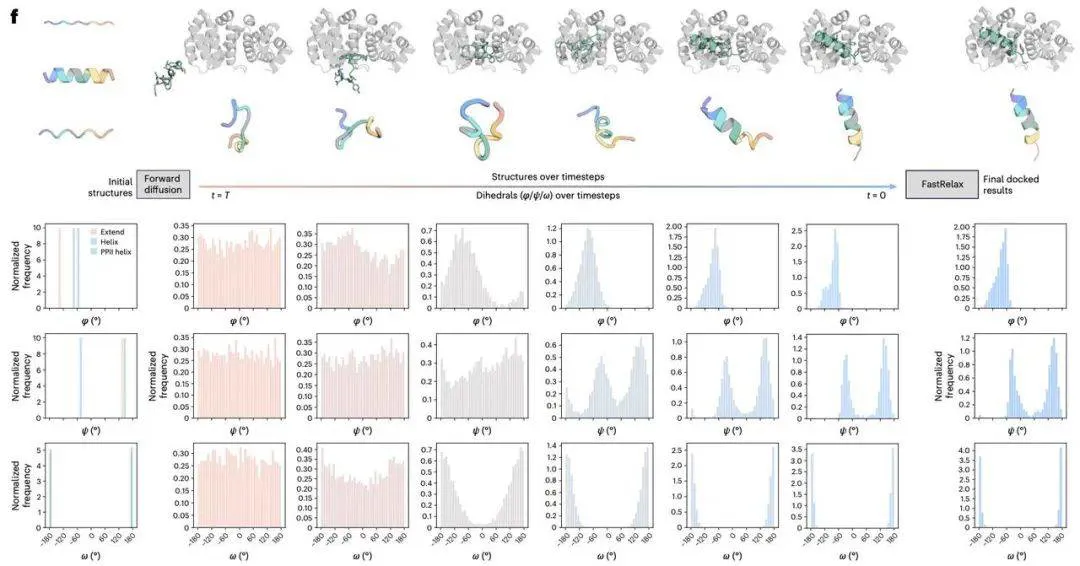
这些策略使 RAPiDock 能在保持优异结构合理性的同时捕捉多层次相互作用细节。
不过,RAPiDock 也存在局限性。一方面,由于一些氨基酸的晶体学数据稀缺,模型训练样本不足,在处理这些氨基酸时精度有所下降,未来可能需要通过扩充修饰氨基酸数据集来解决该问题。
另一方面,由于依赖扩散模型,其迭代式扩散过程会产生较高计算负荷,后续研究可探索通过直接学习构象空间中的平滑轨迹来减少迭代次数。
实验评估
实验表明,RAPiDock 能以极高精度和速度预测蛋白质-多肽对接模式,在多种几何特征上均能获得稳健对接结果,其性能显著优于 AF2Multi_RS、AF3 和 ADCP 等前沿方法。
进一步研究发现,RAPiDock 不仅能成功捕捉结合口袋(binding pockets)的精细结构信息、复现精确的肽结合模式,还可识别蛋白质内多个潜在的肽结合口袋。
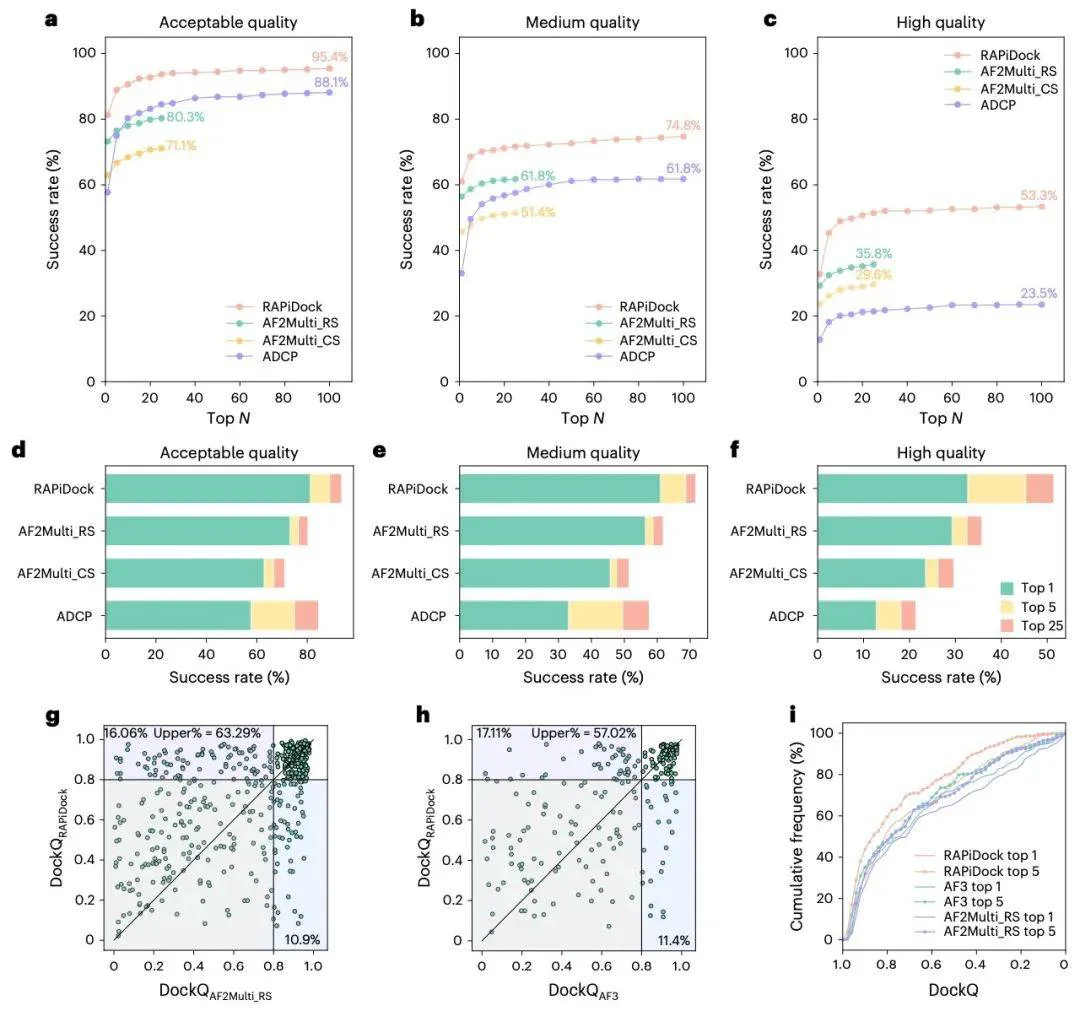
性能测试表明,RAPiDock 在精度与效率上均超越现有最优模型:在 RefPepDB-RecentSet 数据集的 Top-25 预测中,其肽段结合构象预测成功率达 93.7%(较 AF2Multi_RS 提升 13.4%),且单次对接仅需 0.35 秒(速度提升约 270 倍)。
大量案例研究证实,RAPiDock 能处理包括翻译后修饰(PTMs)在内的 92 种残基类型,灵敏识别细微对接模式,成为结构精准的高通量肽段虚拟筛选利器。
总的来说,RAPiDock 以卓越的精度和速度树立了蛋白质-多肽分子对接的新标准,在多重实际应用场景中拓展了高效蛋白质-多肽分子对接的技术边界。
感兴趣的读者可以阅读论文原文,了解更多研究内容。