
编辑丨ScienceAI
人工智能正在重塑精准有机合成的未来,但反应性能预测(数值回归)与合成路线规划(序列生成)之间固有的方法差异,一直是构建统一深度学习架构的挑战。
针对这一难题,上海科学智能研究院(下称上智院)物质科学团队提出了一种名为 RXNGraphormer 的新型深度学习框架。该框架通过整合图神经网络(GNN)与 Transformer 模型,成功实现了对化学反应活性、选择性以及单步正向与逆向合成的精准预测。
这一创新性工作基于超过 1300 万条反应数据的预训练,使模型能够自发学习化学键的变化规律,从而在多项预测任务中达到领先水平。
相关研究成果以《A Unified Pre-trained Deep Learning Framework for Cross-task Reaction Performance Prediction and Synthesis Planning》为题发表于《Nature Machine Intelligence》。

论文地址:https://www.nature.com/articles/s42256-025-01098-4
上智院物质科学领域研究员徐丽成,为论文第一作者、共同通讯作者;上智院AI科学家、物质科学方向负责人曹风雷,上智院首席科学家、复旦大学特聘教授漆远,为论文共同通讯作者;浙江大学博士研究生汤缪炅、上智院物质科学领域研究员安俊逸,为论文共同作者。
统一框架与海量数据驱动的化学键模式学习
RXNGraphormer 的核心突破在于其统一的深度学习架构(图 1),该架构创新性地将反应性能预测(如反应活性和选择性)与单步合成规划(正向与逆向合成)整合至同一系统中。传统方法因这两种任务的内在差异(数值回归与序列生成)而难以实现统一建模,而该框架通过协同设计分子图编码器(GNN)和分子间相互作用编码器(Transformer),并结合创新的预训练策略,成功攻克了这一难题。
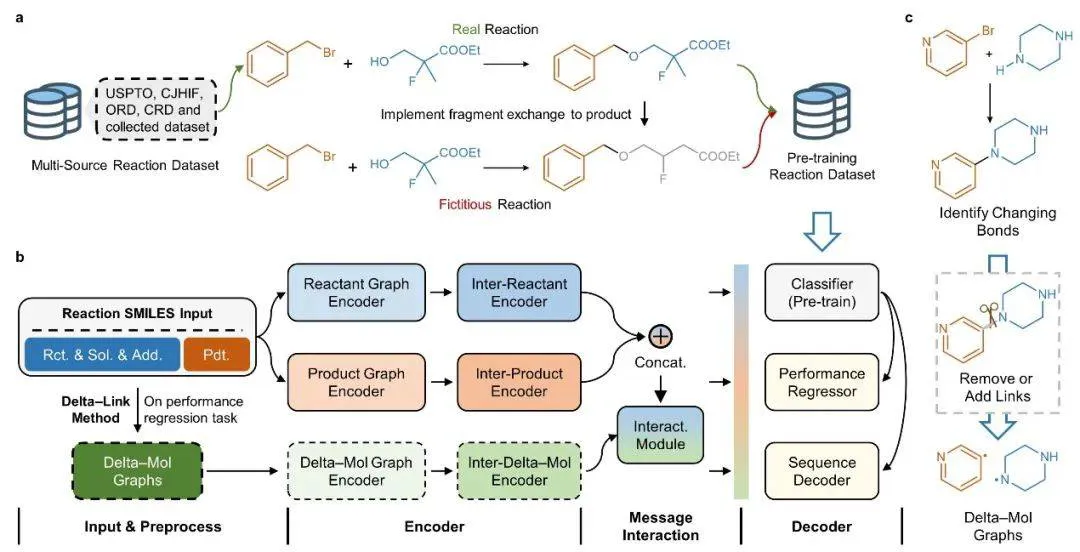
图1:RXNGraphormer 架构设计
研究团队特别开发了一种「片段交换算法」,通过生成虚构反应样本并与真实反应进行对比学习,使模型在预训练过程中自主掌握化学键的断裂与形成模式。这一独特策略不仅使模型无需依赖显式标注即可区分不同反应类型,还能生成具有明确化学意义的嵌入表示。
反应活性与选择性的精准预测
在反应性能预测任务中,RXNGraphormer 通过创新的「delta–link」方法进一步捕捉反应过程中化学键的中间变化,生成「delta–mol」图以增强机制理解。
如图 2 实验表明,该模型在 Buchwald–Hartwig、Suzuki–Miyaura 偶联反应的产率预测中,R² 分别达到 0.971 与 0.876;在区域选择性和对映选择性的预测中,其表现同样优于现有方法。
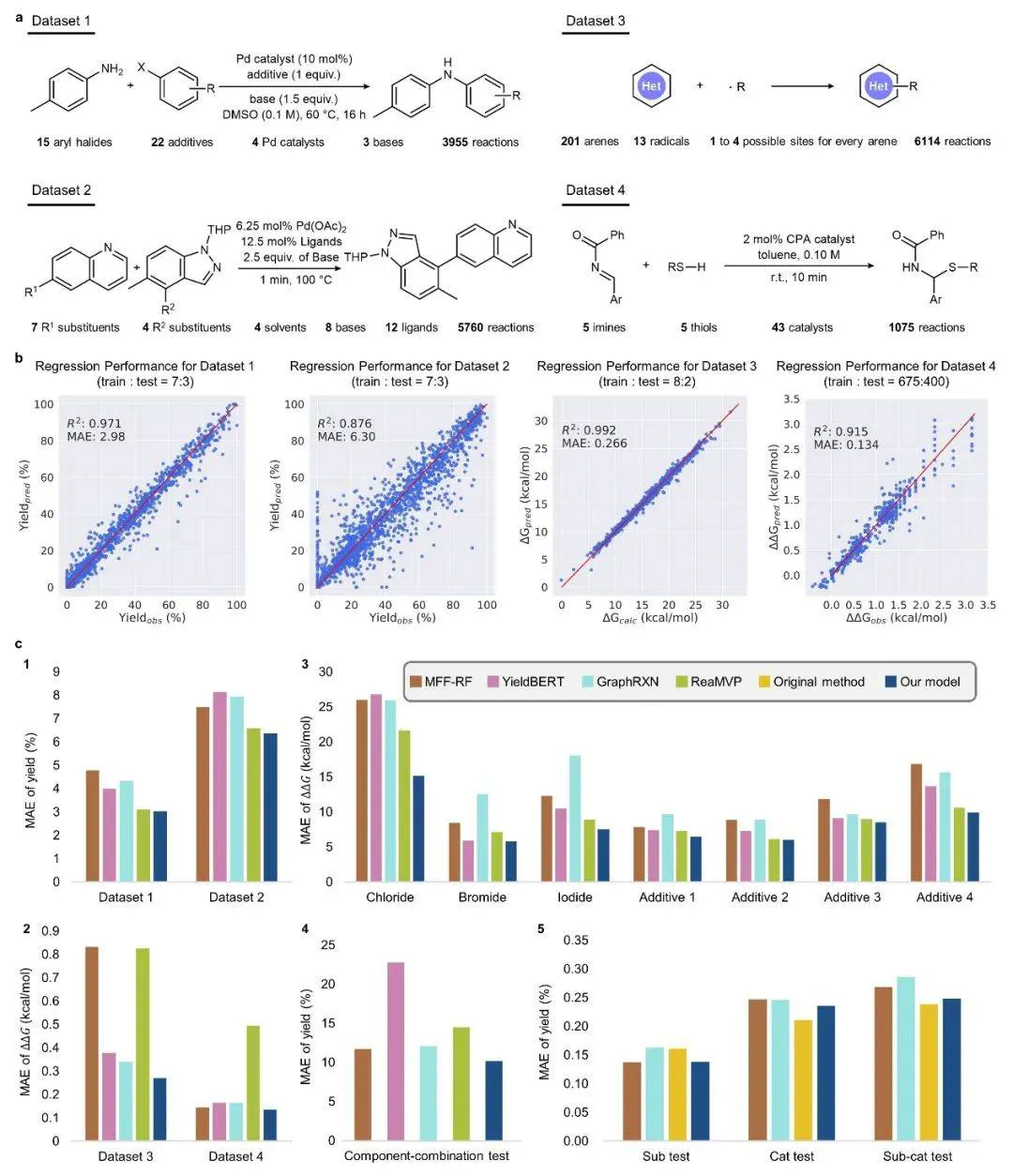
图2 :RXNGraphormer 在反应性能预测任务上的表现
此外,在 3 个更加接近真实反应条件优化场景的额外数据集的测试中,模型同样展现了极高的预测精度与强大的泛化能力。
单步合成规划的卓越表现
在单步逆向合成与正向合成任务中,RXNGraphormer 在 USPTO 系列数据集上刷新了多项无模板方法的记录。例如,在包含百万级反应的 USPTO-full 数据集上,其逆向合成的 Top-1 准确率提升至 47.4%(原无模板法 SOTA 为 45.7%),Top-10 准确率达 71.6%(原无模板法 SOTA 为 67.9%)。
对于涉及立体化学的复杂反应(USPTO-STEREO 数据集),模型在正向合成预测中 Top-1 准确率为 78.2%,显著优于现有方法。
模型自发掌握化学反应分类规律
该研究的突破性进展在于,RXNGraphormer 模型通过整合独特的预训练策略并利用海量反应数据进行训练,展现出对化学反应类型进行自发聚类的卓越能力。这种无监督学习行为表明,模型能够从化学键变化的本质规律中提取深层次的化学认知。在包含 50 类不同化学反应的 USPTO-50k 数据集分析中,模型生成的嵌入空间热图(图 3)直观呈现了各类反应之间的亲疏关系。
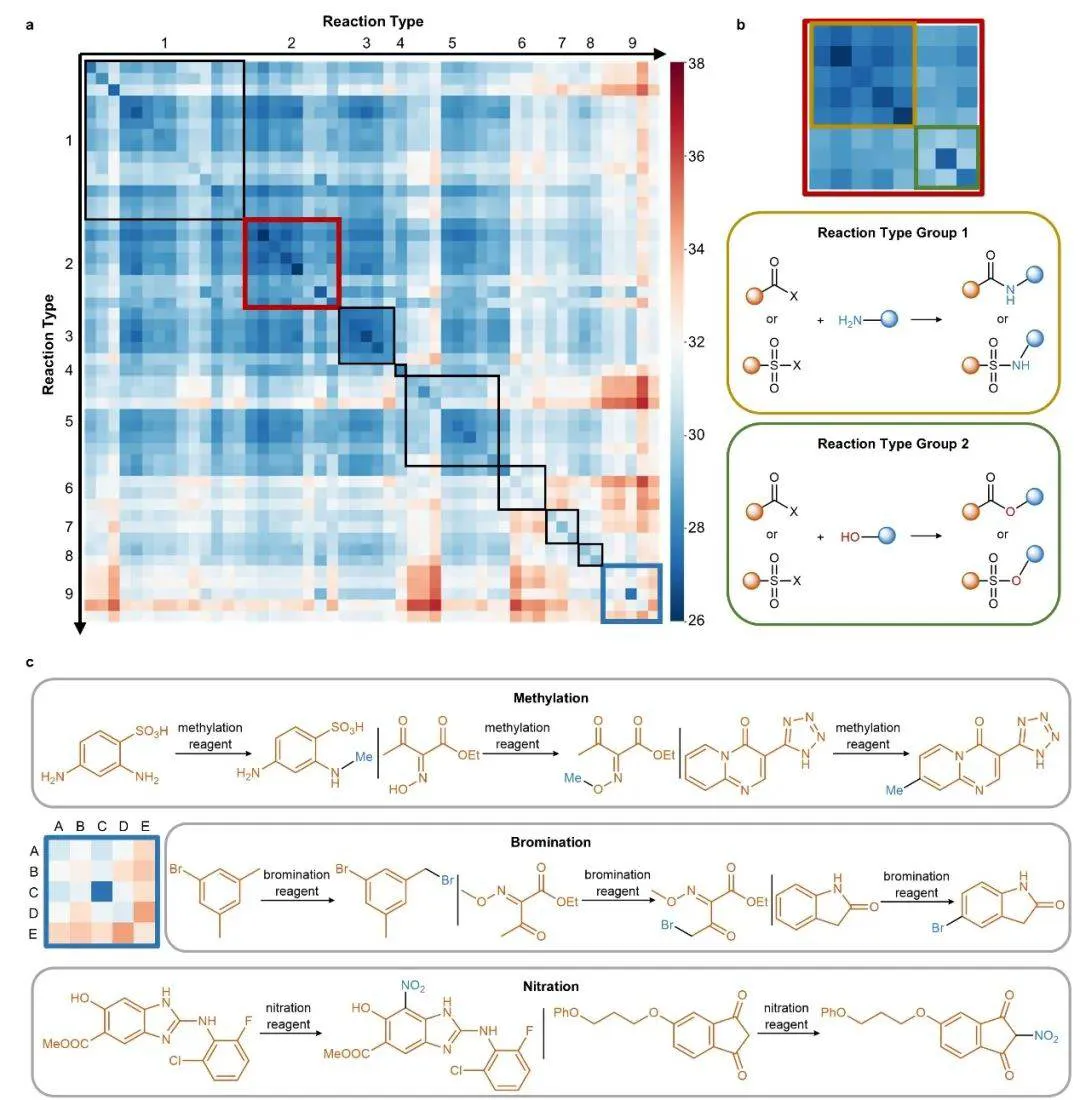
图3:对 USPTO-50k 数据集反应嵌入的距离分析
值得注意的是,这种自发形成的分类模式与传统化学家基于电子效应和键断裂/形成规则的经验分类高度吻合,充分证明该研究设计的预训练策略成功赋予了模型重建化学反应关键特征的能力。
这一发现具有重要的科学价值:首先,它证实了海量反应数据中隐含的键变化模式能够驱动 AI 理解化学反应的深层机制;其次,为反应机理研究提供了全新的数据驱动研究范式。特别值得关注的是,该成果突破了传统依赖人工定义反应分类的局限,展示了 AI 直接从原始数据中提炼化学认知的潜力,为未来机器自主发现化学规律开辟了崭新路径。
RXNGraphormer 的成功标志着化学 AI 领域在统一反应预测框架上的重大进展,其通过数据驱动的化学键规律学习,为精准合成设计与反应优化提供了高效、通用的解决方案。未来,该技术有望加速新药开发与功能分子发现,推动有机合成领域的智能化变革。


