化学

模仿人类直觉的AI,化学本体驱动的反应路径搜索新范式,用知识结构替代暴力搜索
编辑丨&化学反应是化学键的断裂和形成,这是创造新技术的基础。 在发现和开发新的化学反应过程中,时间与精力的双重消耗是研究人员不可避免的头疼问题。 一个分子从反应物到产物,中间可能存在数量巨大的反应路径。

打通计算设计与实验执行闭环,微软、北大、上海交大联合提出科学推理模型QFANG
编辑丨coisini有机合成是分子创新的基础引擎,能够创造出多种先进功能分子。 尽管现代算法能够在计算机中设计数百万种新颖分子,但分子的实际合成仍是主要瓶颈。 这一过程通常需要密集的资源投入,并高度依赖于化学家在多年实践中积累的隐性知识与专业直觉。

化学反应的「全景地图」来了,机器人帮科学家导航高维实验空间
编辑丨&不知道诸位读者在做实验的时候是否经历过一些玄学——明明步骤都是一样的,但就是因为某些奇异的问题,导致实验无法顺利进行下去。 人类化学家只能探索这些流形的一个有限子集,对反应超空间的理解仍然是零碎的 。 产率分布是平滑还是波状?

Nature | 大幅加速多元电催化剂的科学发现,MIT等推出多模态人工智能-机器人平台CRESt
作者 | 论文团队编辑 | ScienceAI近日,美国麻省理工学院李巨团队在国际顶尖学术期刊《Nature》上发表了题为《A multimodal robotic platform for multi-element electrocatalyst discovery》的研究论文。 该工作展示了一种多模态机器人平台 CRESt(Copilot for Real-world Experimental Scientists),通过将多模态模型(融合文本知识、化学成分以及微观结构信息)驱动的材料设计与高通量自动化实验相结合,大幅提升催化剂的研发速度和质量。 论文地址:。

IJCAI 2025 | 化学不是单向路:ChemDual大模型用拆分与重组,优化化学反应与分子逆合成预测
作者 | 论文团队编辑 | ScienceAI化学反应预测和逆合成设计是药物研发中的核心步骤。 近年来,虽然大型语言模型(LLMs)在各领域表现优异,但直接应用到化学合成任务时仍存在两大挑战:数据量稀缺和反应预测与逆合成任务建模割裂。 为了应对数据稀缺问题,湘潭大学林轩副教授课题组联合湖南大学曾湘祥教授、湖南师范大学曾道建教授课题组针对化学反应和逆合成预测,提出了一个新的大模型框架 ——ChemDual。

Nature子刊 | 上智院统一框架RXNGraphormer,实现化学反应多任务精准预测,自发掌握分类规律
编辑丨ScienceAI人工智能正在重塑精准有机合成的未来,但反应性能预测(数值回归)与合成路线规划(序列生成)之间固有的方法差异,一直是构建统一深度学习架构的挑战。 针对这一难题,上海科学智能研究院(下称上智院)物质科学团队提出了一种名为 RXNGraphormer 的新型深度学习框架。 该框架通过整合图神经网络(GNN)与 Transformer 模型,成功实现了对化学反应活性、选择性以及单步正向与逆向合成的精准预测。

AI for Chemistry进入「配置即代码」时代,Chemia一键训练化学AI模型
编辑 | ScienceAI如今,人工智能已成为化学研究不可或缺的工具,广泛应用于化合物性质预测、反应优化和材料设计等领域。 尽管大模型作为当前人工智能领域的主流方向之一,但是基于特征工程的传统人工智能算法依然在许多垂直领域有着难以替代的作用。 目前,垂直科学领域的初学者在面对人工智能建模时,面临诸多挑战,数据处理、模型调参及实验复现等繁琐流程耗时耗力,且易使人望而却步。
性能提高87%,数据需求减70%,天大等开发化学毒性预测模型,登Nature子刊
编辑丨%于化学领域,多物种急性毒性评估构成了化学品分类、标签和风险管理的基础。 传统机器学习模型常因人类毒性数据稀缺(部分端点仅 140 条可用数据)陷入预测困境。 天津大学等联合团队针对传统模型的使用缺陷,开发了最新的条件急性毒性评估(ToxACoL)框架,通过端点关联图建模 伴随双向学习的方式,实现毒性的评估。

科学家用GNN进行不确定性量化,实现高效的分子设计,登Nature子刊
题图来源:AI生成编辑 | 白菜叶在广阔的化学空间中优化分子设计面临着独特的挑战,尤其是在领域转移的情况下保持预测准确性。 在最新的研究中,台湾大学(National Taiwan University)的研究人员整合了不确定性量化 (UQ)、定向信息传递神经网络 (D-MPNN) 和遗传算法 (GA) 来解决这些挑战。 他们系统地评估了 UQ 增强型 D-MPNN 是否能够有效优化广泛、开放的化学空间,并确定了最有效的实施策略。

为矛盾的理论提供统一解释,牛津大学等揭秘非晶硅结构
编辑丨coisini非晶硅(α-Si)是研究最广泛的无序网络固体之一,其结构已经被研究了几十年。 两种主要理论分别基于连续随机网络模型和「类晶体」模型,后者被定义为在保持整体非晶网络的同时,表现出类似于晶体状态的局部结构有序性。 然而,这种局部有序性的程度一直不明确。
11天实现18种广谱抗菌素设计,体外验证成功率94.4%,浙大侯廷军等用LLM方法从头设计AMP
编辑 | 萝卜皮大型语言模型 (LLM) 在化学和生物医学研究中取得了显著进展,可作为各种任务的多功能基础模型。 浙江大学侯廷军、谢昌谕以及南方医科大学姜志辉等课题组组成的联合团队提出了 AMP-Designer,这是一种基于 LLM 的方法,用于快速设计具有所需特性的抗菌肽 (AMP)。 在 11 天内,AMP-Designer 实现了 18 种具有广谱抗革兰氏阴性细菌活性的 AMP 的从头设计。

ByteQC:通往大规模实用化量子化学计算的曙光
编辑 | ScienceAI真实化学体系包含大量的微观粒子,其精确的严格计算需要指数高的复杂度,对这些体系的模拟一直是材料、制药和催化等领域的难点和前沿。 为了解决这一问题,近日字节跳动 ByteDance Research 团队开发并开源了 ByteQC ——基于 GPU 加速的大规模量子化学计算工具集。 该工具集使用强大的 GPU 算力,大幅度加速了常见的量子化学算法,同时结合领域内前沿的量子嵌入方法实现了量子化学「黄金标准」精度下的大规模量子化学体系的模拟。
Transformer革新药物研发:TRACER框架实现反应感知的分子设计与合成优化
编辑 | 2049药物研发周期长、成本高是制药行业面临的重大挑战。 据统计,一个新药从研发到上市平均需要 12 年时间,投入高达 26 亿美元。 为提升研发效率,深度学习在分子生成领域取得了显著进展。

Nature重磅:微软生成式AI材料设计工具,稳定性提升2倍,实验验证误差低于20%!
编辑 | 2049材料创新是推动技术进步的关键驱动力之一。 从 20 世纪 80 年代锂钴氧化物的发现到如今的锂离子电池技术,材料科学的每一次突破都深刻影响着我们的日常生活。 然而,传统的材料发现方法依赖于耗时且昂贵的实验试错过程,而计算筛选方法虽然加速了这一过程,但仍然受限于已知材料的数量。

2024 诺贝尔化学奖得主:「模型幻觉」给我无限创造力
万万没想到,一直备受批评的 AI「幻觉」问题,竟然在科学领域具有极大的应用价值?
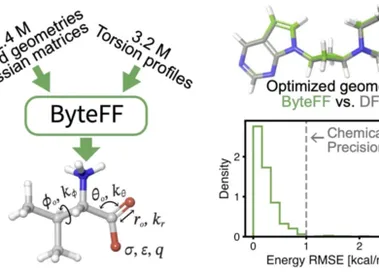
AI赋能传统力场:字节跳动开发高精度通用小分子力场ByteFF
编辑 | ScienceAI小分子力场是药物发现中的重要工具,在计算机辅助药物设计中发挥关键作用。 化学空间覆盖广泛且高效精确的小分子力场将为药物发现奠定可靠的基础。 尽管基于机器学习的 MLFF(如 ANI-2x,MACE-OFF23 等)能够提供非常精确的小分子势能面预测,但它们的训练需要海量数据量,且推理速度较慢,还存在外推场景不确定度大等问题。

AI 驱动科学大爆发!从蛋白质到数学证明,2024 年最值得关注的科技突破
编辑 | ScienceAI2024 年对于 AI for Science 而言,可谓硕果累累:两个诺贝尔奖再度聚焦人工智能与科学的先驱性结合。 其一是诺贝尔化学奖,颁发给了在蛋白质设计与蛋白质结构预测领域做出开创性贡献的 David Baker 博士、John Jumper 博士以及Demis Hassabis 博士;其二是诺贝尔物理学奖,授予了 John J. Hopfield 博士与 Geoffrey Hinton 博士,以表彰他们在人工神经网络及其机学习核心原理方面的奠基性工作。

ScienceAI 2024「AI+材料&化学」专题年度回顾
编辑 | 2049在数字化转型的背景下,人工智能技术正在从根本上改变化学与材料科学的研究范式。 2024年,这场技术革新在多个领域展现其变革力量。 在分子设计领域,基于图神经网络(GNN)和 Transformer 架构的深度学习模型,结合分子动力学模拟,实现了分子性质的精确预测与优化。
资讯热榜
标签云
AI
人工智能
OpenAI
AIGC
模型
ChatGPT
谷歌
DeepSeek
AI新词
AI绘画
大模型
机器人
数据
Midjourney
开源
Meta
微软
智能
用户
GPT
学习
英伟达
Gemini
智能体
技术
马斯克
Anthropic
图像
AI创作
训练
LLM
论文
AI for Science
代码
腾讯
苹果
算法
Agent
Claude
芯片
具身智能
Stable Diffusion
xAI
蛋白质
人形机器人
开发者
生成式
神经网络
机器学习
AI视频
3D
字节跳动
大语言模型
RAG
Sora
百度
研究
GPU
生成
华为
工具
AGI
计算
生成式AI
AI设计
大型语言模型
搜索
亚马逊
AI模型
视频生成
特斯拉
DeepMind
场景
Copilot
深度学习
Transformer
架构
MCP
编程
视觉