编辑丨&
分子性质预测,通常关系到药物与材料研发中的核心要素。现代的科研流程里,图神经网络(GNN)的介入让传统方法更进一步。与此同时,柯尔莫哥洛夫-阿诺德网络 (KAN)提供了更高的表达性、参数效率和可解释性,已经成为多层感知器的强大替代品。
山东大学与新加坡南洋理工大学的研究者对此产生了思考,是否可以结合这两者的优势,将分子预测做得更好、更快、更具可解释性。
他们的研究以「Kolmogorov–Arnold graph neural networks for molecular property prediction」为题,于 2025 年 8 月 11 日刊登在《Nature Machine Intelligence》。

论文链接:https://www.nature.com/articles/s42256-025-01087-7
统一框架 KA-GNN
近年来,得益于诸如 AlphaFold 的模型,药物设计和发现进入了高速发展的新时代。这些基于分子的人工智能模型可以分为两类,即基于分子特征的机器学习和端到端的深度学习。
前者的关键过程是分子特征化,从结构、物理、化学或生物学性质中提取或生成分子特征;后者包括端到端的深度学习模型,使用各种分子表示方法,在分子数据分析和药物设计中得到了广泛应用。
Kolmogorov–Arnold 网络(KANs),基于 Kolmogorov–Arnold 表示定理,在求解偏微分方程中显示出潜力,还在多个应用领域中表现出色,譬如与长短期记忆网络结合,实现了多步时间序列预测的显著改进等。
研究者们所提出的统一框架 KA-GNN,该框架将 KANs 完全整合到 GNNs 的三个核心组件中,包括节点嵌入、消息传递和读出,这种设计既能捕捉低频的全局模式,也能表达高频的局部化结构,从理论上借助 Carleson 与 Fefferman 的收敛性结果给出强近似能力。
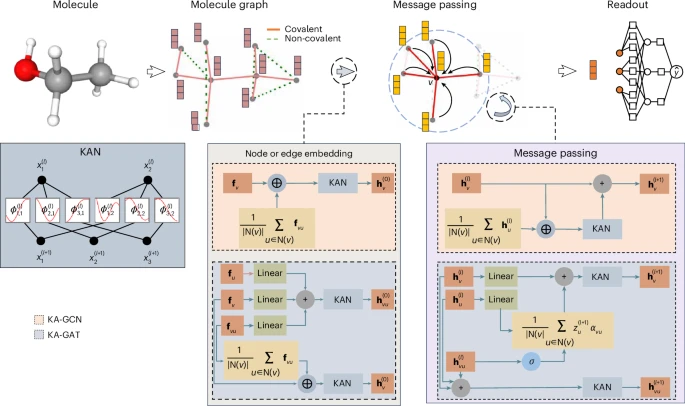
图 1:KA-GNN 模型架构概述。
为了展示团队提出的傅里叶-KAN 模型的强大逼近能力。我们进一步设计了两种变体:KA-GCN(KAN 增强的 GCN)和 KAN 增强的图注意网络(KA-GAT),并在七个基准数据集上的广泛实验验证了 KA-GNN 在准确性和计算效率方面的优越性,确立了其作为非欧几里得数据 GDL 中一个有前途的新范式的地位。
可解释性与具体示例
团队精心准备了 7 个基准数据集,涵盖了生物物理与生理学,多个 GDL 模型与几个其他基于 KAN 的 GNN 架构将会同台竞技。总体结论是:KA-GNN(含 KA-GCN 与 KA-GAT)在准确率和计算效率上均优于传统 GCN/GAT 以及多种 SOTA 图学方法。
在 BBBP 数据集上,KA-GCN 与 KA-GAT 的 AUC 相较基线分别约提升了 7.95% 与 7.68%,并在 ClinTox、MUV 等更具挑战的数据集上也取得显著优势。进一步的消融研究还表明,基于傅里叶变换的 KANs 不仅提高了准确率,还在特征嵌入、消息传递和最终预测阶段也有所改进。
同时,傅里叶-KAN 版本在运行时间和参数规模上表现更优(全局三角基函数使得逼近更紧致、收敛更快),这对大规模分子筛选非常关键。
这些细节在文中都有表格作详细对比,所示结果无不显示着 KA-GNNs 在所有方案中均保持了强大的性能,突显了其对输入尺度变化的稳健性。
除了性能,设计者还重视可解释性:通过对 KA-GAT 的梯度类显著性(saliency)与掩码式 GNNExplainer 分析,模型能自动高亮对预测最重要的原子/键与子图。
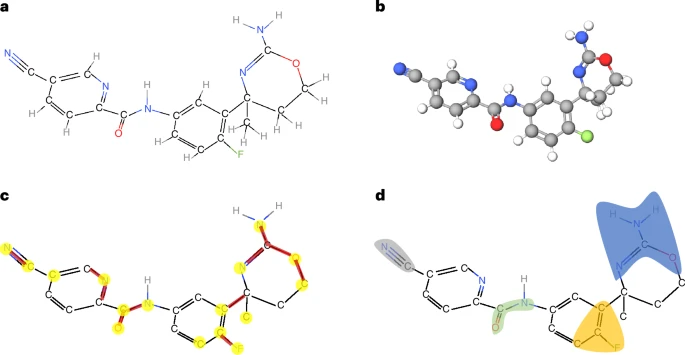
图 2:对某些分子的 KA-GAT 解释。
KA-GNN 的价值在三方面叠加:一是理论支撑,二是工程收益,三是可解释性。这让它既适合传统小分子性质预测,也能扩展到需要考虑更复杂相互作用的预测场景。在工业场景中,模型的效率与可解释性意味着能在更短时间内筛出更可靠的候选化合物,减少「盲投」化学合成的成本。
小结
在如此表现出色的情况下,KA-GNN 仍然可以进一步改进, 尽管显著性图和子图重要性分析已经证明 KA-GNN 能够有效识别关键的分子子结构等,但缺乏将相关傅里叶函数与物理或化学意义联系起来的深入特征重要性分析。
在目前的研究阶段,团队还未使用基于 KAN 的剪枝方法获得更「具有意义」的特征重要性分析结果。不过这终究不是某种数学上的把戏——它能让分子图表现更稳、速度更快、还能告诉你为什么。
未来的工作可以继续把 KAN 与更富物理先验的图构造结合,并在更大、更多模态的数据集上检验泛化性。别忘了:模型能告诉研究者「可能的化学信号」,但最终的药理学、毒理学验证仍要回到实验台——这是机器与人合作、而非机器独断的最好证明。


