编辑丨&
随着单细胞技术的发展,研究者可以在数以万计的细胞中同时测量多个基因或分子标记,并且通过遗传、化学或环境干扰(perturbation)引入实验变量,深入理解细胞反应机制。
这种类型的数据不仅体量巨大,而且结构复杂,不同实验条件、不同细胞类型和干扰策略之间的差异,使得传统的分析工具难以有效覆盖整体流程。现有方法大多只针对单个任务,或者专注于某种类型的环境干扰,而缺乏一个能够统一管理、分析和解释各种单细胞扰动实验的平台。
考虑到现有的生物背景,德国慕尼黑亥姆霍兹中心(Helmholtz Center Munich)与慕尼黑工业大学等(Technical University of Munich)提出了一个基于 Python 的模块化框架 pertpy,可用于分析大规模单细胞扰动实验。
相关研究内容以「Pertpy: an end-to-end framework for perturbation analysis」为题,于 2025 年 12 月 31 日发布在《Nature Methods》。

论文链接:https://www.nature.com/articles/s41592-025-02909-7
端到端的框架
单细胞技术的进步,尤其是 Perturb-seq、CROP-seq 等高通量扰动技术的出现,让科学家能够以前所未有的规模进行“细胞实验”。他们可以同时敲除成千上万个基因,或施加数百种药物,并在单细胞分辨率下观察结果。这为系统性理解基因功能、药物机制和疾病通路提供了革命性的窗口。
但这种实验常被数据的庞大数量级冲垮。现有的工具,如 MUSIC、ScMAGeCK 等,只擅长处理特定类型的扰动或解决单一问题。而为了解决扩展性与通用性的框架缺失问题,pertpy 团队给出了自己的看法。
团队的解决方案并非简单地堆积功能,pertpy 的设计哲学是模块化、互操作与可扩展。它包含分析单一和组合扰动的方法,涵盖多种扰动数据类型,包括遗传敲除、药物筛选和疾病状态。该框架设计灵活,提供 100 多个可组合且互作的分析功能,组织成模块,进一步简化后续的解释和可视化。
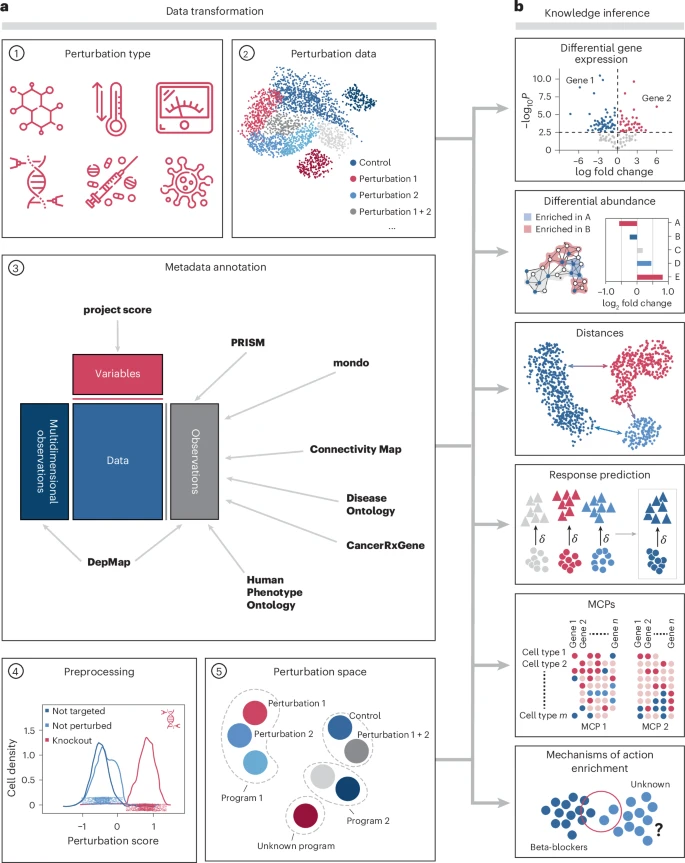
图 1:pertpy 框架的模块。
团队表示,尽管设计中 pertpy 主要设计用于探索遗传改造、药物治疗等扰动,但其效用也扩展到多种扰动环境,包括未应用实验扰动的多种疾病状态。所有这些功能通过 JAX 库实现 GPU 加速,其速度相较于原始实现有数量级提升。
首先,框架通过数据转化,将引导 RNA(gRNA)分配给细胞。接下来,它会处理诸如技术变异、其他单细胞特异性质量控制问题等不受欢迎的混杂因素。
经过严格的质量控制后,pertpy 开始对细胞系本体或药物本体进行扰动注释处理,并用来自癌症依赖地图的额外元数据丰富扰动。而为了迎接扰动数量增加带来的挑战,pertpy 提供了多种不同方式来学习生物学上可解释的扰动空间,这些方法不同于细胞的个体主义视角,而是每个扰动生成一个单一嵌入,汇总细胞反应。
场景验证
为验证 pertpy 学习有意义扰动空间的能力,团队分析了最初由 Norman 等人公开发布的 CRISPRa 筛查数据集。,包含 111,255 个 K562 细胞的单细胞转录组,经历了 287 次单基因和基因对扰动。
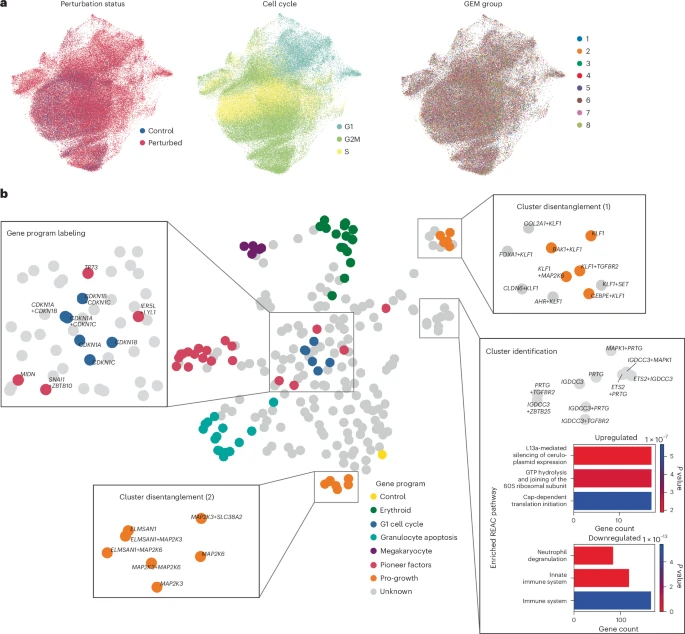
图 2:通过 pertpy 的扰动空间流水线,学习组合 CRISPRa 扰动 scRNA-seq 数据中的统一扰动空间。
团队测试了多种针对微扰的处理策略,并利用基于多层感知子(MLP)的判别器分类器,将剩余细胞的归一化基因表达投射到扰动空间中。
结果表示,所有策略产生的微扰空间相似。这表明对于该数据集,不依赖基于微扰特征的单元过滤方法更为可取。
而面对复杂的微扰实验的发现流程, pertpy 同样以极高的效率分析了包含 172 个细胞系和 13 种药物治疗的数据集。这只需要几个步骤:注释、可视化、比较分析。这其中还允许用户将其细胞系的 RNA 谱与已建立的公开数据集进行比较,从而提供快速的质量控制功能。
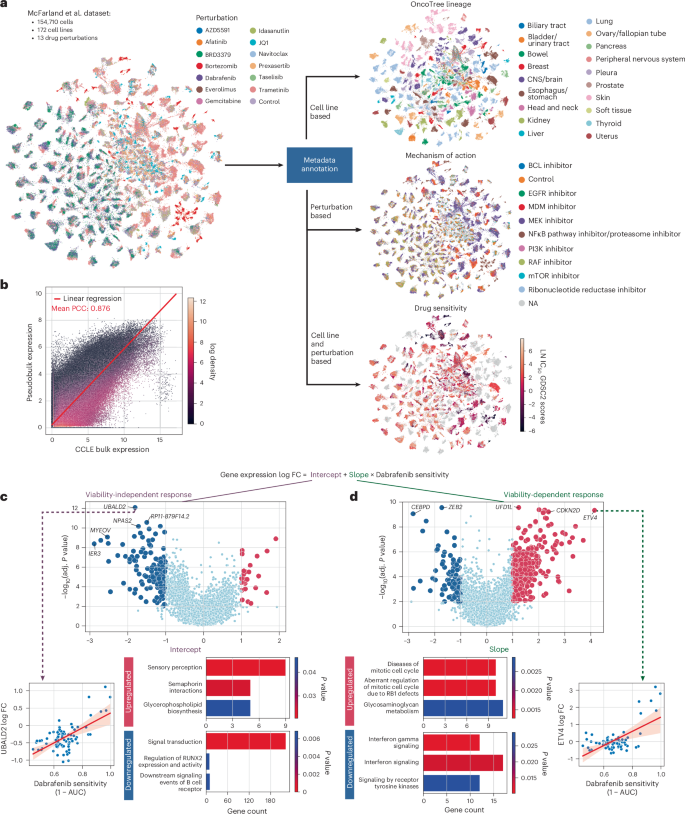
图 3:scRNA-seq 药物筛选数据中存活性相关反应特征的解卷积。
可扩展的单细胞扰动分析
作为一款开源的分析工具,pertpy 将过去分散的单细胞 perturbation 分析方法整合到一个结构化、可重复、易扩展的框架中。它极大地降低了领域门槛,为构建大规模扰动图谱奠定了基础。
Pertpy 不仅为研究者提供了工具链,还为未来算法的开发和集成奠定了基础,是单细胞 perturbation 研究数据层面解决方案的重要一步。它提供的丰富距离度量和分析模块,正是评估这些模型预测是否具有生物学意义的标尺。