分子

MaskMol:知识引导的分子图像自监督学习框架,破解活性悬崖识别难题
作者丨论文团队编辑丨ScienceAI在药物发现领域,一个长期存在的挑战是活性悬崖(Activity Cliffs)现象,即两个分子结构高度相似但生物活性却存在显著差异,然而,由于传统方法在识别活性悬崖时,会出现表征坍塌的问题,因此仍然难以准确捕捉悬崖分子的细微差别。 近日,湖南大学等团队在《BMC Biology》上发表了一项研究《MaskMol: knowledge-guided molecular image pre-training framework for activity cliffs with pixel masking》,这项研究开发了名为 MaskMol 的知识引导分子图像自监督预训练框架,该框架能通过细粒度知识引导的像素掩码策略进行准确的活性悬崖估计。 活性悬崖与传统模型的局限分子性质预测是药物研发的核心环节,其核心假设是结构相似则性质相似(图 1 左)。
10/20/2025 11:56:00 AM
ScienceAI

奥本大学等利用深度学习模拟展现「越拉越牢」的反直觉分子动力学,揭开逆锁键的受力瞬间秘密
编辑丨%日常生活中,物品收到过大的力后常常会断裂开——就像被掰开的意大利面。 但在分子世界中,有些结合键却表现得像「唱反调」:越拉越牢。 这类奇特的相互作用被称为逆锁键(catch bonds)。
9/23/2025 11:52:00 AM
ScienceAI

Meta 发布巨型 AI 化学数据集 OMol25及通用模型 UMA
Meta 近日重磅发布了其迄今为止规模最大的 AI 驱动化学开放数据集 OMol25,并同步推出了用于预测分子和材料化学性质的通用人工智能模型 UMA(Universal Atom Model)。 这两项创新成果旨在加速药物研发、电池材料开发和催化剂研究等关键领域。 据 Meta 介绍,OMol25数据集包含了超过1亿次高精度的分子计算数据,其规模远超此前任何公开的同类数据集。
5/16/2025 10:01:05 AM
AI在线

科学家用GNN进行不确定性量化,实现高效的分子设计,登Nature子刊
题图来源:AI生成编辑 | 白菜叶在广阔的化学空间中优化分子设计面临着独特的挑战,尤其是在领域转移的情况下保持预测准确性。 在最新的研究中,台湾大学(National Taiwan University)的研究人员整合了不确定性量化 (UQ)、定向信息传递神经网络 (D-MPNN) 和遗传算法 (GA) 来解决这些挑战。 他们系统地评估了 UQ 增强型 D-MPNN 是否能够有效优化广泛、开放的化学空间,并确定了最有效的实施策略。
4/10/2025 1:11:00 PM
ScienceAI
剑桥团队革新RNA速率分析:AI算法突破基因动态追踪
编辑丨&RNA 速率模型利用剪接和未剪接的 RNA 计数中包含的时间信息来推断转录动力学,但现有的速率模型通常依赖于粗略的生物物理简化或数值近似来求解基础常微分方程(ODE)。 英国剑桥大学主导的团队提出了 cell2fate,它允许以完全贝叶斯方式求解生物物理学上更准确的模型。 通过将 RNA 速率解决方案分解为模块,cell2fate 在 RNA 速率和统计降维之间建立了生物物理联系。
3/20/2025 2:12:00 PM
ScienceAI
原子级精确,David Baker团队开发RFdiffusion变体对抗体进行从头设计
编辑 | 萝卜皮抗体在现代医学中发挥着核心作用,但目前还没有完全在计算机中设计与特定表位结合的新型抗体的方法。 相反,抗体发现目前依赖于动物免疫或随机文库筛选方法。 华盛顿大学 David Baker 团队证明,结合使用微调 RFdiffusion 网络的计算蛋白质设计与酵母展示筛选,可以生成抗体可变重链(VHH)和单链可变片段(scFv),以原子级精度结合用户指定的表位。
3/18/2025 11:58:00 AM
ScienceAI

基于药效团与GPT从头生成分子,北大开发TransPharmer助力药物发现
编辑 | 萝卜皮深度生成模型推动了药物发现,但生成的化合物通常结构新颖性有限,限制了药物化学家的灵感。 为了解决这个问题,北京大学的研究人员开发了 TransPharmer,这是一种生成模型,它将基于配体的可解释药效团指纹与基于生成预训练 Transformer(GPT)的框架相结合,用于从头生成分子。 TransPharmer 在无条件分布学习、从头生成和药效团约束下的骨架构建方面表现出色。
3/11/2025 12:16:00 PM
ScienceAI
用「完形填空」的思维生成蛋白-分子复合物,西湖大学等发布CBGBench助力药物设计
编辑 | ScienceAI本文作者为西湖大学 Haitao Lin,Yufei Huang, 深势科技 Guojiang Zhao,Zhifeng Gao,及华盛顿大学 Odin Zhang 等;通讯作者为 Stan Z. 被 ICLR2025 接受为 spotlight paper。 原文链接::(Structure-Based Drug Design, SBDD)旨在生成能够与目标蛋白结合的潜在药物分子,并在生成式 AI 技术的推动下得到了极大加速。
2/27/2025 3:52:00 PM
ScienceAI

「定制化」结合蛋白质,几何深度学习方法加速开发精准药物,登Nature
编辑 | 萝卜皮蛋白质是生命的基础,具有多样的生物功能,如输送氧气、传递化学信号和防御病原体。 其分子表面的特异性决定了其功能,这一特性被用于药物开发,通过设计分子与特定蛋白质结合来改变其结合方式,甚至开发「分子胶」来治疗疾病。 奥地利科学院(ÖAW)的 Michael Bronstein、瑞士洛桑联邦理工学(EPFL)的 Bruno Correia 等,率先使用了一种名为「MaSIF(molecular surface interaction fingerprinting)」的几何深度学习架构,用于设计具有所需分子表面特性的新蛋白质。
1/26/2025 7:23:00 PM
ScienceAI
英矽智能在Nature 子刊发表最新研究,利用量子-经典混合模型设计新型KRAS抑制剂
近期,英矽智能与加拿大多伦多大学共同主导的一项研究首次展示量子计算和人工智能在变革药物发现流程方面的潜力。 在这项研究中,科学家将量子计算模型与经典计算模型和生成式人工智能相结合,通过对庞大数据集的训练、生成和筛选,探索更广泛的化学可能性,发现靶向“不可成药”癌症驱动蛋白KRAS 的新颖分子。 这项研究也得到了圣裘德儿童研究医院等多方研究机构的支持,相关研究成果于1月22日发表于Nature Biotechnology。
1/23/2025 11:26:00 AM
新闻助手
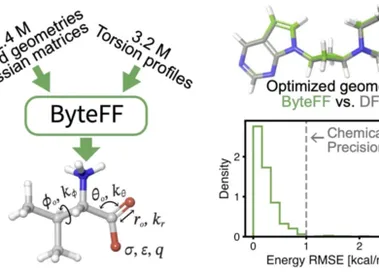
AI赋能传统力场:字节跳动开发高精度通用小分子力场ByteFF
编辑 | ScienceAI小分子力场是药物发现中的重要工具,在计算机辅助药物设计中发挥关键作用。 化学空间覆盖广泛且高效精确的小分子力场将为药物发现奠定可靠的基础。 尽管基于机器学习的 MLFF(如 ANI-2x,MACE-OFF23 等)能够提供非常精确的小分子势能面预测,但它们的训练需要海量数据量,且推理速度较慢,还存在外推场景不确定度大等问题。
1/16/2025 11:49:00 AM
ScienceAI

分子特性预测新框架来了!浙大侯廷军团队、匹兹堡大学联合提出跨通道学习,各大基准表现亮眼
编辑 | 萝卜皮可靠的分子特性预测对于各种科学研究和工业应用(例如药物研发)至关重要。 然而,由于数据稀缺,加上物理化学和生物特性与传统分子特征化方案之间的高度非线性因果关系,使得开发稳健的分子机器学习模型变得异常复杂。 匹兹堡大学(University of Pittsburgh)与浙江大学侯廷军团队合作开发了一种多通道预训练框架,可以稳健学习利用化学知识。
1/9/2025 5:38:00 PM
ScienceAI

远程 “闻” 香,数字嗅觉公司 Osmo 用 AI 技术实现气味“传送”
一家名为 Osmo 的“数字嗅觉”公司宣布,已成功利用 AI 技术分析一个位置的气味,并在无需人工干预的情况下在其他地方复制它。 气味隐形传态使用传感器收集气味,并通过气相色谱质谱仪 (GC / MS) 进行分析。 然后,数据被传输到专门的分子打印机,它可以合成和组合精确再现气味所需的化学物质。
11/4/2024 11:18:37 AM
汪淼
原子、分子、复合物级性质预测均最佳,清华分子预训练框架登Nature子刊
编辑 | KXAI 已广泛用于药物发现和材料设计中的分子筛选。当前的自监督分子预训练方法往往忽略了分子的基本化学特性和物理原理。为了解决这个问题,来自清华大学的研究人员提出了一种称为分数去噪(Fractional denoising,Frad)的分子预训练框架,以学习有效的分子表示。通过这种方式,噪声变得可自定义,允许纳入化学先验,从而大大改善分子分布建模。实验表明,Frad 始终优于现有方法,在力预测、量子化学特性和结合亲和力任务中,取得最先进的结果。改进的噪声设计提高了力准确性和采样覆盖范围,这有助于创建物理一
9/20/2024 12:08:00 PM
ScienceAI

打开AI黑匣子,「三段式」AI用于化学研究,优化分子同时产生新化学知识,登Nature
编辑 | KXAI 工具的强大功能,令人难以置信。但如果你试图打开引擎盖并了解它们在做什么,你通常会一无所获。AI 常常被视为「黑匣子」。对于化学来说,AI 可以帮助我们优化分子,但它无法告诉我们为什么这是最佳的——重要的特性、结构和功能是什么?近日,伊利诺伊大学厄巴纳-香槟分校(UIUC)的一个跨学科研究团队打开了黑匣子,研究人员通过将 AI 与自动化学合成和实验验证相结合,找到了 AI 所依赖的化学原理,从而改进用于收集太阳能的分子。研究找到了比现有稳定四倍的捕光分子,同时给出了使其保持稳定的重要见解 ——这是
9/4/2024 2:19:00 PM
ScienceAI

Nature子刊,优于AlphaFold,全原子采样,一种预测肽结构的AI方法
编辑 | 萝卜皮深度学习方法推动了生物分子结构单态预测的重大进展。然而,生物分子的功能取决于它们可以呈现的构象范围。对于肽来说尤其如此,肽是一类高度灵活的分子,参与多种生物过程,作为治疗手段备受关注。多伦多大学的 Philip M. Kim 和 Osama Abdin 开发了 PepFlow,这是一种可转移生成模型,它能够从输入肽的允许构象空间中直接进行全原子采样。研究人员在扩散框架中训练模型,然后使用等效流进行构象采样。为了克服广义全原子建模的成本过高,他们模块化了生成过程并集成了超网络来预测序列特定的网络参数。
7/8/2024 6:40:00 PM
ScienceAI
分子100%有效,从头设计配体,湖南大学提出基于片段的分子表征框架
编辑 | KX分子描述符广泛应用于分子建模,但在 AI 辅助分子发现领域,缺乏自然适用、完整且「原始」的分子表征是一个挑战,影响 AI 模型的性能和可解释性。在使用先进的自然语言处理(NLP)方法解决化学问题时,会出现两个基本问题:(1)什么是「化学词」?(2)如何将它们编码为「化学句子」?近日,湖南大学研究团队提出了一种灵活的、基于片段的多尺度分子表征框架 t-SMILES 的框架来解决第二个问题。该框架使用 SMILES 类型的字符串描述分子,并且可以将基于序列的模型作为主要生成模型。t-SMILES 具有三种
7/5/2024 2:44:00 PM
ScienceAI
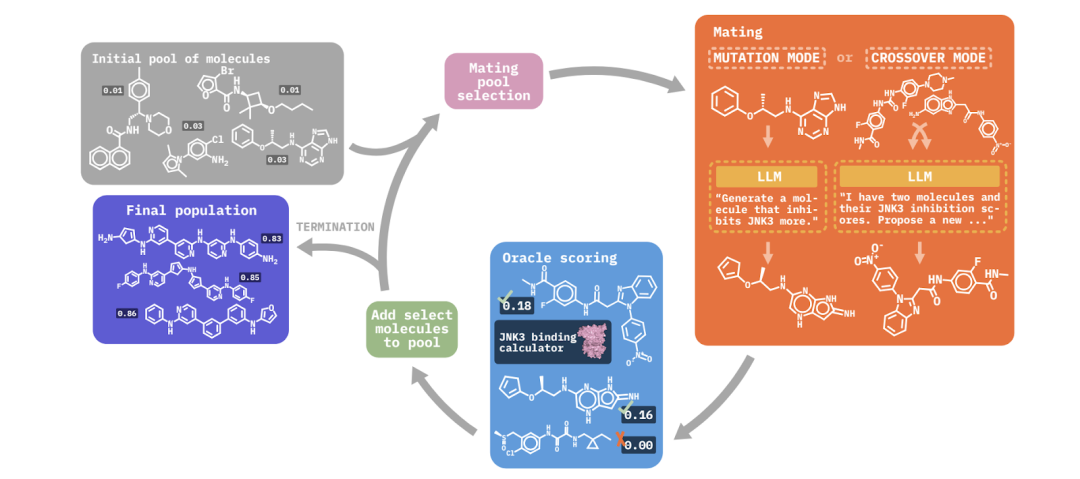
击败25个分子设计算法,佐治亚理工、多伦多大学、康奈尔提出大语言模型MOLLEO
作者 | 佐治亚理工学院王浩瑞编辑 | ScienceAI分子发现作为优化问题,因其优化目标可能不可微分而带来显著的计算挑战。进化算法(EAs)常用于优化分子发现中的黑箱目标,通过随机突变和交叉来遍历化学空间,但这会导致大量昂贵的目标评估。在这项工作中,佐治亚理工学院、多伦多大学和康奈尔大学研究者合作提出了分子语言增强进化优化(MOLLEO),通过将拥有化学知识的预训练大语言模型(LLMs)整合到进化算法中,显著改善了进化算法的分子优化能力。该研究以《Efficient Evolutionary Search Ov
7/1/2024 4:14:00 PM
ScienceAI
资讯热榜
标签云
AI
人工智能
OpenAI
AIGC
模型
ChatGPT
DeepSeek
谷歌
AI绘画
大模型
机器人
数据
AI新词
Midjourney
开源
Meta
微软
智能
用户
GPT
学习
技术
智能体
马斯克
Gemini
Anthropic
英伟达
图像
AI创作
训练
LLM
论文
代码
算法
AI for Science
苹果
Agent
Claude
腾讯
芯片
Stable Diffusion
蛋白质
开发者
xAI
具身智能
生成式
神经网络
机器学习
3D
人形机器人
AI视频
RAG
大语言模型
研究
百度
Sora
生成
GPU
工具
华为
计算
字节跳动
AI设计
AGI
大型语言模型
搜索
生成式AI
视频生成
场景
DeepMind
特斯拉
深度学习
AI模型
架构
亚马逊
Transformer
MCP
编程
视觉
预测