
作者 | 论文团队
编辑 | ScienceAI
原子级模拟为材料性质的预测与虚拟筛选提供了重要手段,其核心在于准确描述体系的势能面(PES)。相比于传统 PES 的计算所依赖的方法——从头算量子化学方法(如密度泛函理论 DFT)与经验力场而言,机器学习原子间势函数可以更好权衡精度与效率,通过从 DFT 数据中学习原子间相互作用,对 PES 进行准确构建。
近年来,随着数据和模型规模的扩大,基于数百万 DFT 计算训练的基座势能(foundation potentials, FPs)进一步展现出跨化学空间的泛化潜力,已被广泛用于声子谱预测、相图构建、催化筛选与分子动力学模拟等下游任务,为多尺度材料建模奠定了新的基础。
然而,绝大多数 FPs 依然是在大量低精度 GGA/GGA+U 的 DFT 泛函数据上训练的,随着我们对于下游任务提出更高的模拟精度的需求,经低精度数据预训练的模型亟待通过对于高精度数据(如 r2SCAN、HSE06 泛函)的迁移学习,构造为精度更高的模型。
为应对这一跨泛函迁移挑战,来自加州大学伯克利分校(UCB)的黄旭,邓博文,钟佩辰及 Gerbrand Ceder 教授等,在名为 CHGNet 的基座势能框架内分析了这一跨泛函由低精度数据到高精度数据迁移学习问题的挑战及解决方案。
该研究以「Cross-functional transferability in foundation machine learning interatomic potentials」为题,于 2025 年 10 月 21 日刊登于《npj Computational Materials》。

论文链接:https://www.nature.com/articles/s41524-025-01796-y
基座势能在当前训练数据的局限及跨泛函迁移的挑战
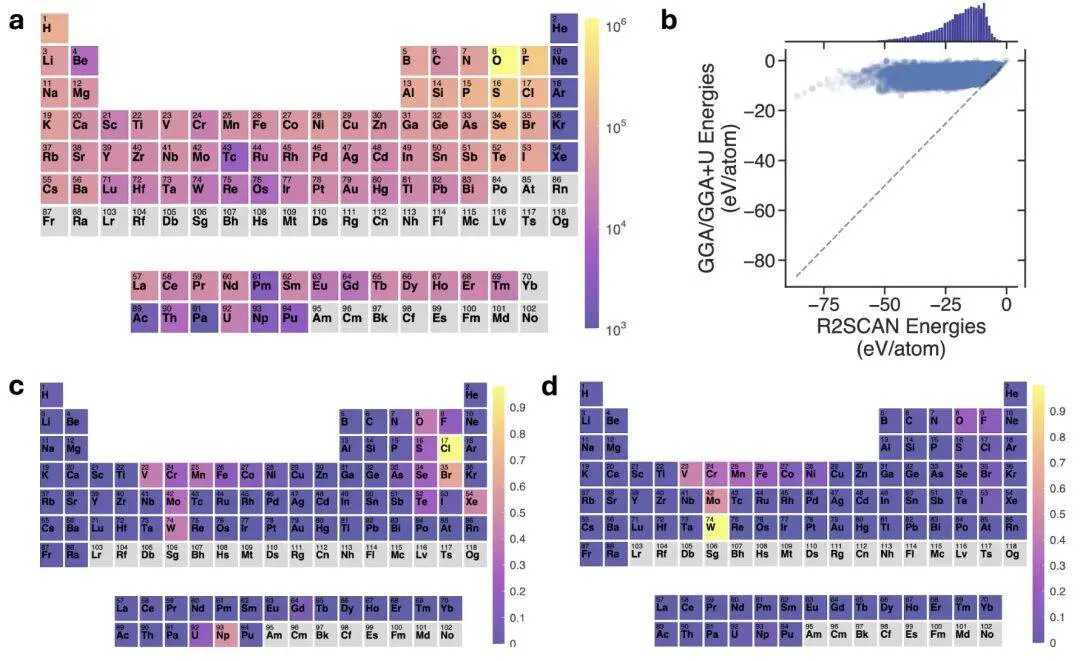
目前用于晶体材料的基座势能(FPs)训练的数据集主要是 GGA 和 GGA+U 水平的 DFT 泛函数据组成。
首先,GGA/GGA + U 泛函在不同化学键环境下的迁移能力较低,整体计算精度较低。其次,为减少 GGA 中的自相互作用误差而施加的 Hubbard U 校正,本质上是半经验性的且具有非普适性。「最优」U 值并没有明确的定义,线性响应法等方法表明,这样的最优 U 值通常依赖于具体体系。
然而,GGA/GGA + U 的 FP 数据集在生成时,对每种元素都使用相同的 U 值,而不考虑局部环境或形式价态。再次,我们通常采用一种粗粒度的方案将 GGA 与 GGA + U 混合以构建 FPs 训练集。这样的混合方式可能引发问题,例如在这些训练数据之间移动时,原子间势能可能会出现数百 meV 的突跃,这是不利于平滑势能面构建的。
在多精度 DFT 数据集之间实现显式或隐式的跨泛函可迁移性的主要策略有三种:迁移学习(transfer learning)、多精度学习(multi-fidelity learning)以及混合多精度训练(mixed multi-fidelity training)。
这三种方法有各自的优势及挑战,其中,迁移学习指的是先在大规模的低精度数据集上对一个大型神经网络进行预训练,然后将该网络的预训练权重用于初始化在更小规模的高精度数据集上的机器学习任务。这种方法在计算和数据利用上都非常高效。然而,如果不同精度数据集之间的相关性不够强,迁移学习的效果就会变差,甚至可能导致性能下降,这种现象被称为「负迁移」(negative transfer)。
MP-r2SCAN 数据集
研究团队构建了一个来自 Materials Project 数据库的 r2SCAN 弛豫轨迹解析而成的 r2SCAN 数据集(MP-r2SCAN),用于高精度训练任务,包含 34,927 个材料 ID 和 238,247 个结构。与 MPtrj 数据集(包含 145,923 个材料 ID 和 1,580,395 个结构)相比,MP-r2SCAN 的数据规模显著更小。
如下图图 b 所示,r2SCAN 与 GGA/GGA + U 的总能量分布在不同的数值范围上。从 GGA/GGA + U 到 r2SCAN 的能量偏移量在 0–70 eV/atom 的量级,这远大于势函数能量精度(约 30 meV/atom),这表明这些 r2SCAN 能量标签若没有合适的参考或归一化处理,是无法直接有效迁移使用的。
使用不同原子参考能量的迁移学习及基准测试
在一些 FPs 模型内部,会将总能量预测分为两部分: (由原子参考能量所得)和
(由原子参考能量所得)和 (图神经预测)。
(图神经预测)。 是由每个结构组成代表的行向量
是由每个结构组成代表的行向量 和原子参考能向量
和原子参考能向量 的点积得到(见下图)。其中,
的点积得到(见下图)。其中, 可以根据训练数据提前进行线性拟合得到。
可以根据训练数据提前进行线性拟合得到。
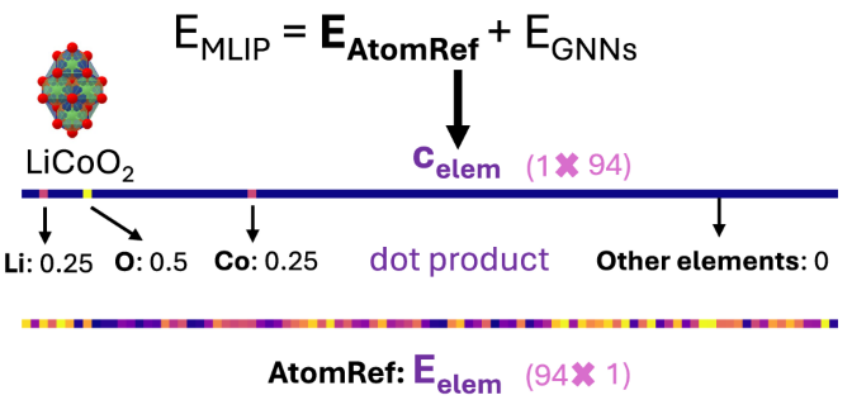
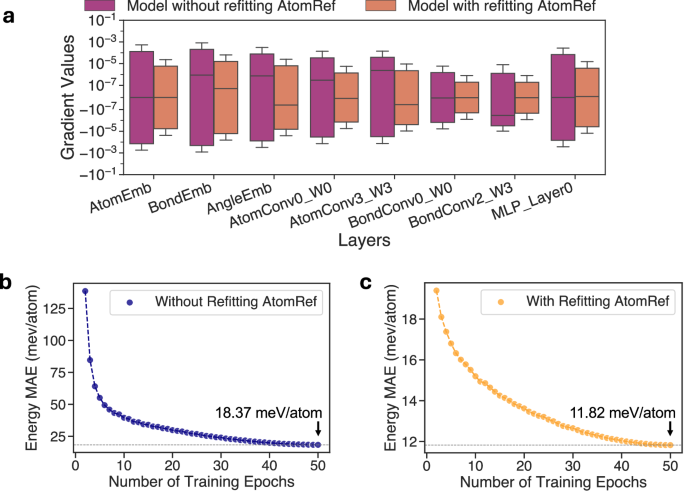
对于在具有基于 GGA/GGA + U 总能量拟合的 AtomRef 的势函数 FP 上进行的跨泛函迁移学习,可以重新拟合该 FP 的 AtomRef(用基于 r2SCAN 能量拟合得到的 AtomRef 替换原有的基于 GGA/GGA + U 的 AtomRef),从而将 FP 迁移前后图神经网络学习的能量预测部分对齐到相近的数值尺度,并提升预训练与微调数据集之间的相关性,克服迁移学习中「负迁移」的挑战。实际上,Pearson 相关系数 ρ 从未修正的 GGA/GGA + U 与 r2SCAN 数据集之间的 0.0917,提高到了在分别减去各自 AtomRef 后的 r2SCAN 能量与 GGA/GGA + U 能量之间的 0.9250。此外,此种方法也减少了微调开始阶段梯度过大导致的训练不稳定性。由下图可以观察到,不重新拟合 AtomRef 的方法的初始梯度幅值至少比重新拟合 AtomRef 的方法大一个数量级,且重新拟合 AtomRef 后,模型的训练过程更加稳定且可靠。
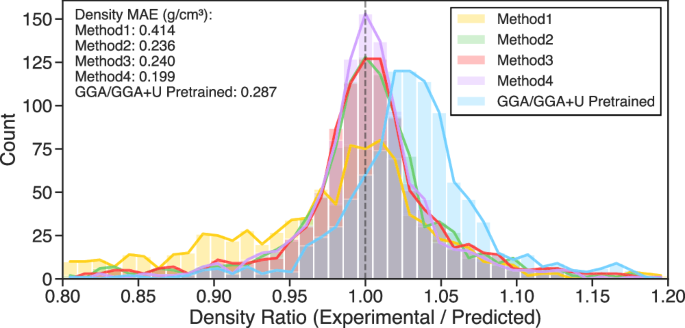
在基准测试方面,该研究将重新拟合原子参考能量进行迁移学习的方法和其他几个迁移学习方法以及从头用 r2SCAN 数据训练(无迁移学习)的方法进行了对比,得到在能量、原子间作用力、应力、磁矩、热力学稳定性(形成能、分解能)、密度预测方面都更准确可靠的 FPs。如下图所示,方法 4(即重新拟合原子参考能量的迁移学习)在密度预测(与实验密度数据对比)方法显著优于其他方法。此外,实验值与预测值之比的分布显示,基于 GGA 预训练的 CHGNet 模型更倾向于低估密度,而方法 4 所得到的分布相比其他方法更紧密地集中在理想值(比值 = 1)附近。上述结果表明,从 GGA 向 r2SCAN 的迁移学习方法是有效的,并且使用 r2SCAN 数据、通过方法 4 训练得到的模型在实际体积与密度预测中具有更高的精度潜力。
除了从 GGA/GGA+U 到 r2SCAN 泛函数据迁移学习,团队也研究了从 GGA/GGA+U 到卤化物范德华数据集和 HSE06 数据集的迁移学习(见论文中的补充信息),证明了这种迁移方法的普适有效性。
扩展定律
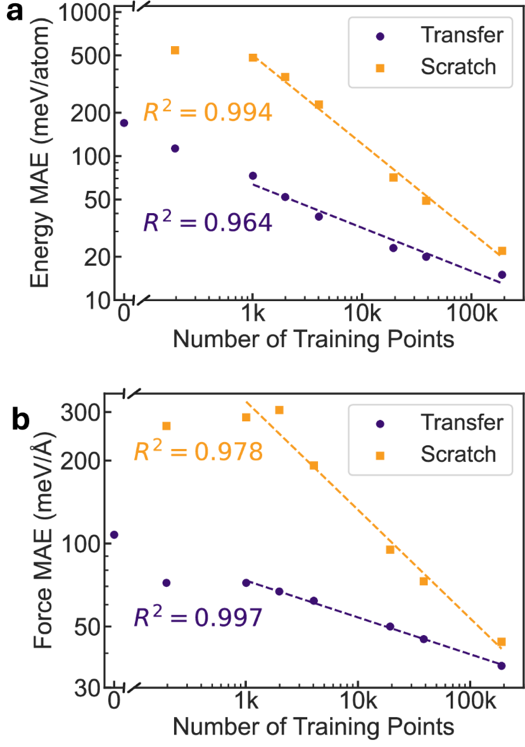
为评估重新拟合原子参考能量的迁移学习方法的数据效率提升,研究团队进行了扩展定律研究(下图)。线性拟合结果表明,无论是从头用 r2SCAN 数据训练(无迁移学习)还是迁移学习(蓝色),都表现出线性扩展规律的行为。仅使用 1K 个高精度数据点进行迁移学习,就能超越在超过 10K 个高精度数据点上从头训练的模型性能,这意味着通过 GGA 预训练步骤获得了超过十倍的数据效率提升。有趣的是,研究团队观察到,即使在包含 24 万个结构的完整 MP-r2SCAN 数据集上,迁移学习相较于从头训练的优势仍未饱和,表现出出色的数据效率。
总结与展望
研究团队通过在 MP-r2SCAN 数据集上对不同 TL 方法进行得到在能量、原子间作用力、应力、磁矩、热力学稳定性(形成能、分解能)、密度预测方面的基准测试,证明了 FPs 迁移学习中重新拟合原子参考能量的重要性及此方法的有效性。并且,通过比较具有和不具有低精度数据集预训练的扩展定律,研究表明了得当的迁移学习方法可以实现显著的数据效率提升。


