编辑丨%
看病就医,医生会先进行诊断,在了解病症的基础上,给出治疗建议。传统药物设计也遵循类似的逻辑——建立在对病原体或疾病机制的了解之上。
现在,AI 带来了一种出乎意料的突破:就像无需看病直接吃药一样,它能够设计出粘附并分解体内有害蛋白质的小分子,即使科学家完全不清楚这些蛋白质的结构与形态。
这是一个很直观的想法:如果把目标蛋白序列末端的「肽结合段」掩蔽起来,微调模型以重构结合区,或许就能直接根据目标序列条件生成能结合该靶点的肽,而无需结构输入。美国宾夕法尼亚大学(University of Pennsylvania)与杜克大学(Duke University)等据此提出了一种基于靶序列设计的全新线性肽配体设计模型 PepMLM。
他们的研究成果以「Target sequence-conditioned design of peptide binders using masked language modeling」为题,于 2025 年 8 月 13 日刊登在《Nature Biotechnology》。

论文链接:https://www.nature.com/articles/s41587-025-02761-2
从序列直接设计结合子
在过去的几年里,深度学习通过注意力机制的应用,彻底改变了自然语言处理(NLP),特别是在蛋白质等其他语言模型中的应用。研究团队之前所开发的 PepPrCLIP 模型首先从潜在空间中采样自然肽候选者,然后通过对比模型进行筛选以确定目标序列特异性。
但很遗憾的是,一种纯粹从头开始、基于目标序列的结合基序设计算法还尚未开发出来。
为了实现这一目标,团队引入了 PepMLM 算法,基于掩码语言模型,建立在 ESM-2 的基础上。他们把目标蛋白序列与其已知肽结合段拼接,在训练时把肽段全部掩码,任务就是重构这部分序列。
通过专注于肽区域的完整重建,PepMLM 成为一种完全基于序列、条件于靶标的从头设计结合剂工具,为开发更有效的、针对构象多样的蛋白质的治疗性结合剂铺平了道路。
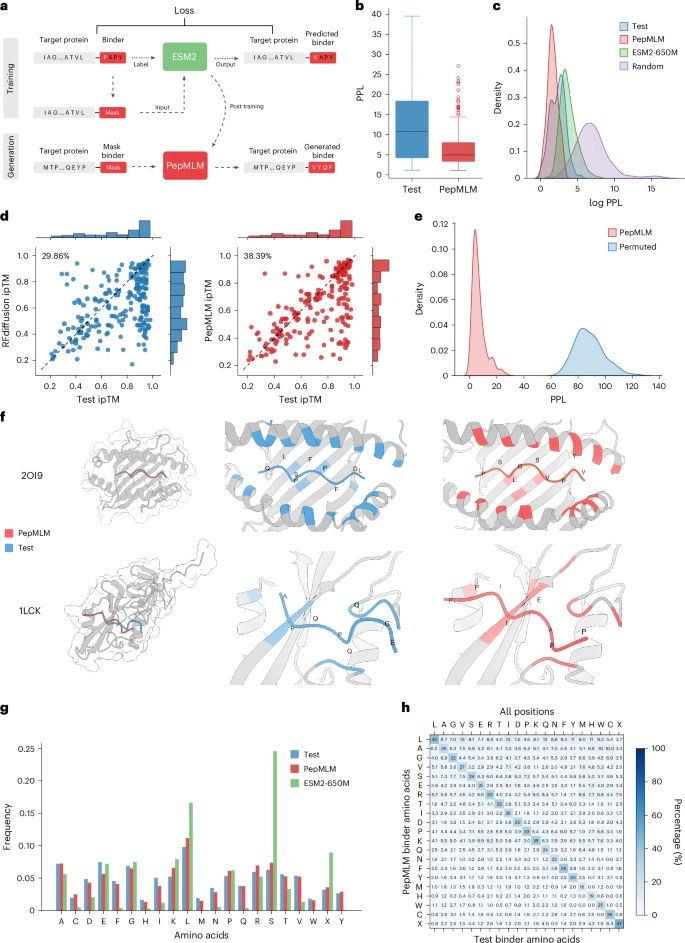
图 1:PepMLM 模型的概述与评估。
训练数据经聚类与去冗余后,构成了约 10,000 条训练样本与 203 条测试样本(binder 长度 ≤ 50,目标序列 ≤ 500),并用 pseudo-perplexity(PPL)作为对候选肽的置信度评分。生成时采用 greedy 或 top-k(最终选 k=3)采样以平衡多样性与置信度。该做法把「目标条件化 + 掩码重构」作为设计策略的核心。
分布分析显示,PepMLM 在低 PPL 区域与真实结合剂的分布非常接近,ESM-2 模型本身和随机设计的结合剂所观察到的分布偏移表明,PepMLM 可以通过 PPL 评分区分结合剂和非结合剂。
基准测试与实验对比
在 203 个测试目标上,PepMLM 的 PPL 分布与真实已知结合肽更为一致,能把「可信」结合子集中在低 PPL 区域,这一点通过与 AlphaFold-Multimer 的联合评估得到支持。
在氨基酸组成水平上,PepMLM 设计的序列与测试结合体的氨基酸分布高度一致,而 ESM-2 则表现出强烈的偏向丝氨酸(S)、亮氨酸(L)等。这些都表明在微调后,PepMLM 更好地捕捉了蛋白质-肽相互作用中的天然氨基酸偏好。
对于测试结合体中的每个位置,团队分析了 100 个设计结合体中相应位置的氨基酸类型,在所有位置和接触位置观察到 69.2% 和 68.4% 的氨基酸特异性变化。除了这些,在在训练分布之外的泛化中,PepMLM 并没有表现出对高同源性的依赖,而且能够很好地泛化到未见过的蛋白质底物上,激励对多种与疾病相关的靶标进行实验表征。
接下来,团队把 PepMLM 生成的肽嵌入到 uAb 等可诱导降解构架中进行细胞实验验证,展示了两个方向的证据链:
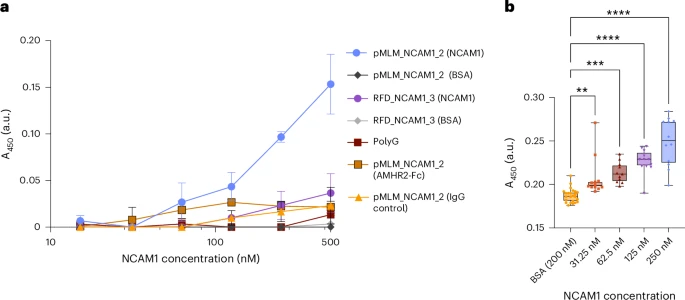
图 2:体外 PepMLM 设计的肽结合子的实验验证。
在体外/细胞结合与降解实验中,PepMLM 生成的肽针对多种靶点(包括 NCAM1、AMHR2,以及 Huntington 相关靶点 HTT 等)表现出序列特异性结合并能诱导蛋白水平下降。PepMLM 仅从目标序列生成有前景的结合体候选物,并且成功率高于当前最先进的结合体设计模型。
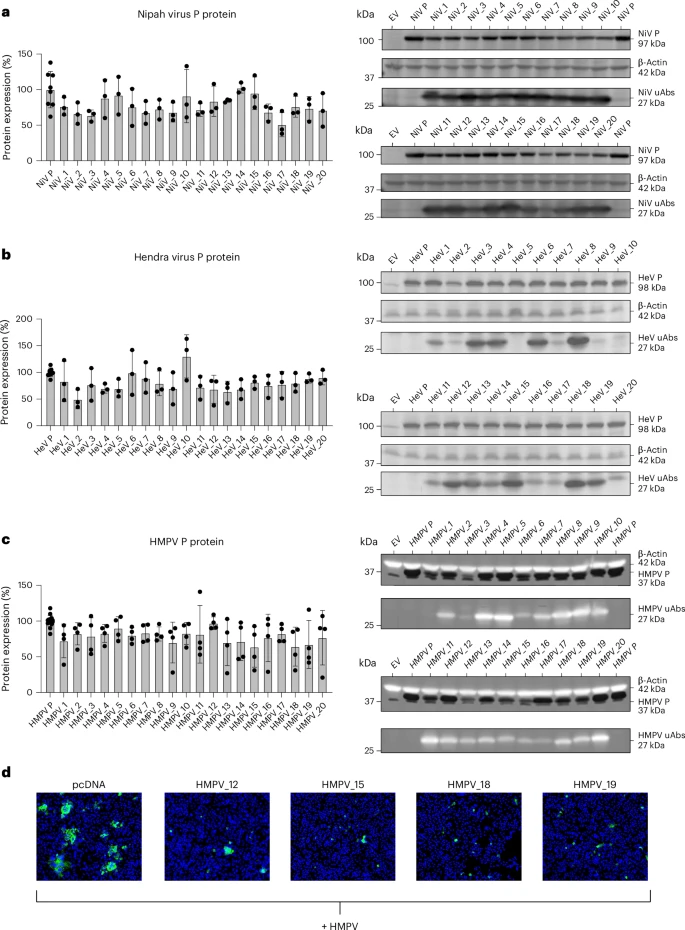
图 3:体外筛选源自 PepMLM 的抗病毒 uAb。
另一方面,针对三种高危病毒(NiV、HeV、HMPV)磷蛋白的筛选,实验里通过 PepMLM 设计了 20 款 uAb,后续的筛查显示约 37 个 降解剂 在 20%–49% 的范围内降低目标蛋白表达(总体命中率 ~63%),并有若干在感染模型中将磷蛋白近乎完全清除的候选。
整体而言,实验命中率与计算层面的 hit rate 呈一致趋势,证明 PepMLM 的设计可转化为生物功能。
小结
PepMLM 是 ESM-2 的微调版本,采用简单的掩码-解掩码方案,提供了一种设计线性肽结合物的简便框架。研究团队表示,PepMLM 不是标准的生成序列模型,与传统的自回归或离散扩散和流匹配模型相比有所不同。
尽管使用的表述更为简约,但团队依旧有力地证明了 PepMLM 在体外、体内以及治疗相关的情境下都能生成强大的结合物设计。 它把「掩码式语言建模」应用到目标条件化肽设计上,做到无需结构输入即可生成、优先排序并在细胞中验证候选肽。
总体来看,这是一条「从序列到候选再到功能验证」的可行路径,尤其适合那些结构不可得或高度可变的难以设计药物靶点群体。