编辑丨&
传统监测抗菌耐药(AMR)常靠序列比对:有相似参考就能识别,没有就白瞎了。这种「参考驱动」的策略一方面可靠,另一方面也很脆弱——数据库里没有代表性的参考序列,就很容易把真正的耐药蛋白漏掉(假阴性)——尤其是当耐药蛋白多样性超出已知的参考范畴。
为此,巴西圣保罗大学(University of São Paulo)等的研究者训练了一个卷积神经网络(CNN)以区分抗微生物耐药性蛋白和非耐药性蛋白,将其命名为 DeepSEA。它的出发点很简单:让模型学会直接从原始氨基酸序列里识别耐药「信号」,不要一直靠找近亲。
经过研究后的 CNN 能够对九种蛋白质类别进行分类,并且能够将它们与非耐药蛋白区分开来,召回率(真阳性/相关元素)超过 0.95。
相关研究内容以「DeepSEA: an alignment-free explainable approach to annotate antimicrobial resistance proteins」为题,于 2025 年 9 月 1 日发布在《BMC Bioinformatics》

论文链接:https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-025-06256-4
把「训练样本」和「非耐药样本」都摆干净
研究以 NCRD95(从 CARD、SARG、ARDB 等整合并用同源搜索扩展的数据库)做为抗性蛋白的主训练源,并限制相似度为 95% 的版本来平衡代表性与重复性
非耐药类(NonR)则从 SwissProt 的经人工审校子集里筛出约 191,535 个细菌蛋白,并用 CD-HIT 限制相似度,最终从中随机抽取与训练集无明显比对的约 4600 条样例作为 NonR,以避免类内不均衡导致模型学偏。
整个数据预处理对比对阈值、类别累积曲线与子类剔除都做了细致控制,目的是把「训练集的偏见」降到最低。
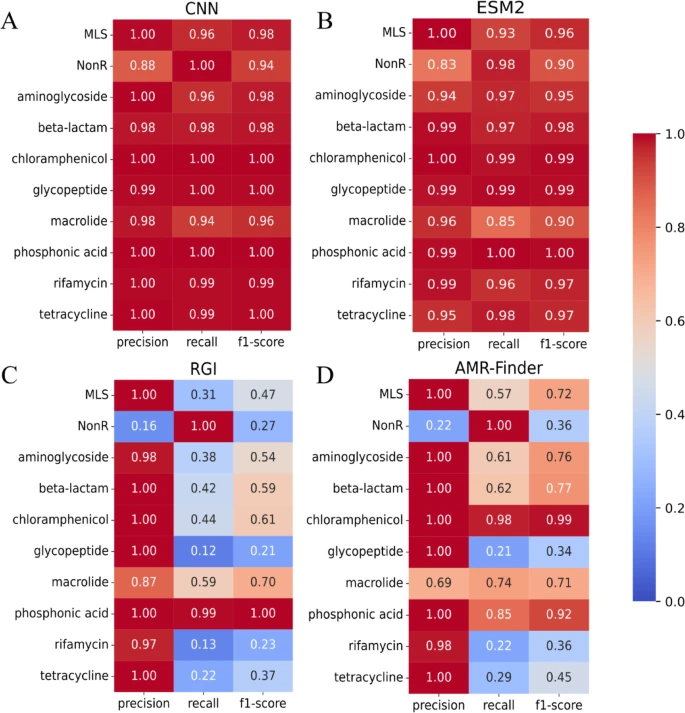
图 1:保留测试集上的分类报告。
DeepSEA 在多类耐药注释任务上表现非常亮眼,基于深度学习的方式在总体召回(recall)在文章中报告均显著高于传统基于比对的方法(recall > 0.95),也就是说它能把更多真实的耐药蛋白「拽出来」而不是漏过去。
RGI 与 AMRFinderPlus 在检测对糖肽类(glycopeptides)耐药蛋白时分别把 88% 和 79% 的这类蛋白误判为「非耐药」,而 DeepSEA 在同类任务中仅错判 8 个 β-内酰胺耐药蛋白为非耐药,这直接说明了 DeepSEA 在减少假阴性方面的优势。与复杂、预训练巨模型 ESM2 的比较显示,两者性能相当,但 DeepSEA 的架构更轻、可解释性更好。
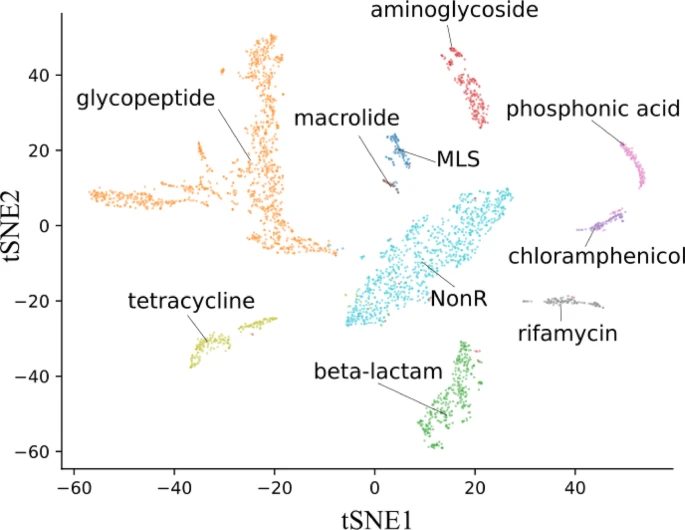
图 2:CNN 类集群。
在对外部数据库 NDARO 的拓展验证中,DeepSEA 也表现稳健:NDARO 中的 5959 条耐药蛋白中 98% 被 DeepSEA 正确分类;对于那些不对齐(no-hit)的 233 条蛋白,DeepSEA 只错判了 42 条。研究者据此判断:只要训练集中包含该耐药「类」的示例,模型就能较好地泛化到序列相似度低的样本上。
把 CNN 的「黑盒」掰开看内部火花
为了便于理解模型的内部表征,研究还把全局平均池化层的向量做 t-SNE 降维,并在 holdout 集中呈现出按功能类别聚簇的清晰图景,这既是模型「学到类间差异」的证据,也为生物学家提供了探索新子类的线索。
DeepSEA 的直接用法很明确:在元基因组或未注释基因组的注释流程中,遇到「无比对」的蛋白序列时,传统工具常判 「非耐药」,但 DeepSEA 可以补上一把火,把那些结构/功能上虽与参考不同但却能导致耐药性的蛋白识别出来。
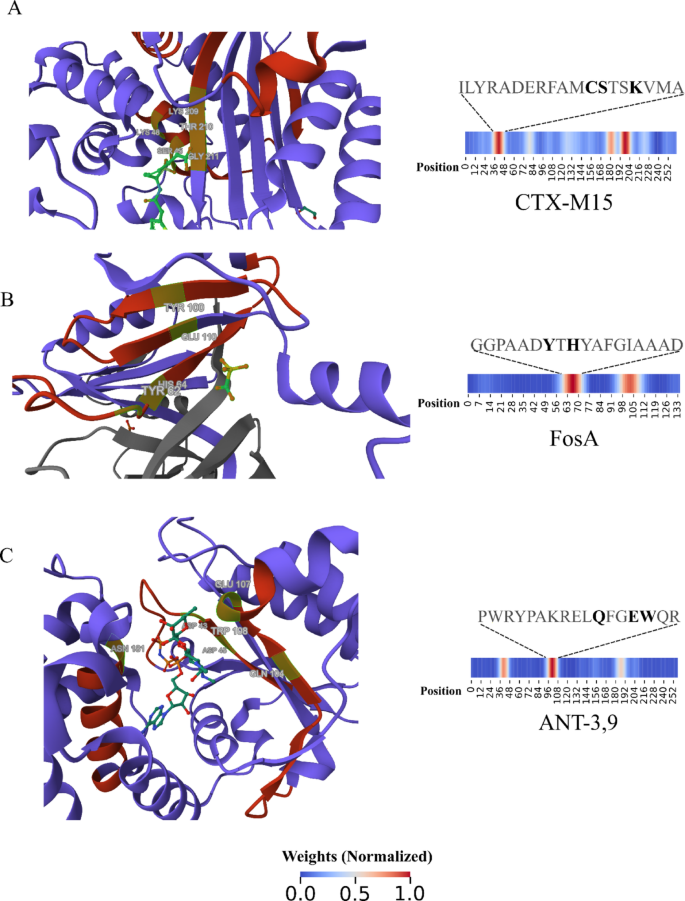
图 3:CNN 模型可解释性。
研究者建议将 DeepSEA 用作「再注释」(reannotation)工具,或者作为注释流水线里对比对方法的补充,从而降低漏报率。代码与工具已开源放在 GitHub,方便整合到现有管道。
当然,DeepSEA 也有其局限性:如果训练集中根本没有某个耐药子类的代表,模型仍难以识别;非耐药类(NonR)内部信息极其杂乱,会导致对该类的精确率下降;模型可解释性虽然做了较多工作,但并不能替代结构生物学或功能实验的最终判定。
结论
扼要地说,DeepSEA 提供了一个不依赖严格序列比对、既能高召回又便于解释的 AMR 蛋白注释方案。它在减少假阴性上尤其有效,对低相似度样本也能较好泛化;同时,它也将可解释性做成了工程化的输出,方便生物学家把「AI 的判断」映射回已知的功能域或活性位点。
它不是想取代比对工具,而是把注释生态从「只看相似度」扩展为「相似度+表征学习+可解释性」的混合范式。若把 DeepSEA 同现有注释链条并用,短期内能显著提高耐药蛋白的检出率;长期来看,把它和结构/功能实验联动,能把「预测的可信度」再往上推一档。