编辑丨&
在经典物理中,电子穿过金属导线的路径就像一场拥挤的通勤:无数次碰撞、阻力与能量损失。但在某些量子材料中,它们似乎找到了捷径——弹道输运(ballistic transport):电子从一端射入,几乎不发生散射地滑行到另一端。
这意味着电流几乎无损,就像高速公路上只有一辆车,笔直驶向出口。
自 1970 年 Landauer 建立输运模型以来,科学家一直用这一理论描述「理想条件下」的弹道电子:电子只能从通道两端注入或流出,路径光滑、无杂质、无散射。问题是——现实的量子材料远非理想。
来自德国 Jülich 研究中心(Forschungszentrum Jülich)与亚琛工业大学(RWTH Aachen)等的研究团队决定,重新描绘电子的真实旅程。他们提出了一种名为 Distributed Current Injection 的新模型,用于探测真实条件下的弹道电子。
该研究以「Distributed Current Injection into a One-Dimensional Ballistic Edge Channel」为题发表在《Physical Review Letters》上。

论文链接:https://journals.aps.org/prl/abstract/10.1103/l47r-plxq
从理想状态走到现实
这一支研究团队抛弃了 Landauer 的一个关键假设:
「电流只能从导体的端点注入与流出。」
相反,它允许电子在通道的任意位置进入或离开。这意味着模型能描述更真实的情况——电子与外部电极、缺陷或探针发生弱耦合时的行为。
研究者以此计算出一个新特征:在这样的系统中,沿着通道测得的电压分布不再线性,而是出现独特的非均匀曲线。
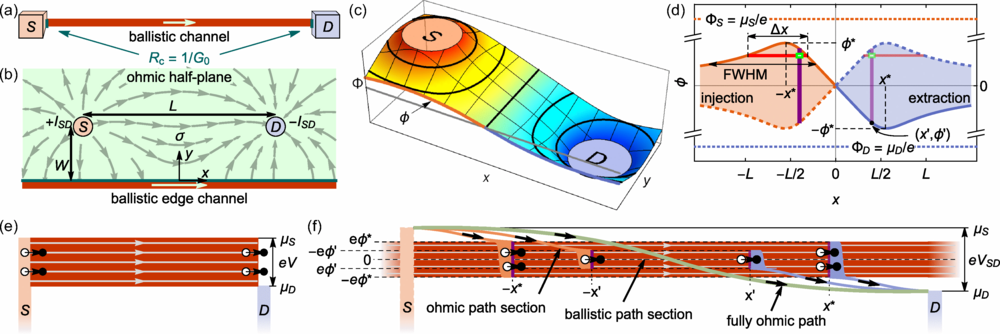
图 1:一维弹道边缘通道的分布式注入。
这一结论非常关键:如果实验测得这种电压形态,就可以直接判定电子确实以弹道方式流动。换句话说,该研究团队给出了「如何识别真正的弹道」的实验判据。
以拓扑绝缘体的边缘通道为例,这些材料的电子仅能沿样品边界传播,内部完全绝缘,因此是观察弹道输运的理想平台。在新模型中,研究团队引入了分布式电流源项,允许电流密度在空间上连续变化。他们解出了修正后的泊松方程与电位分布函数,得到一个带位置依赖项的电压解。
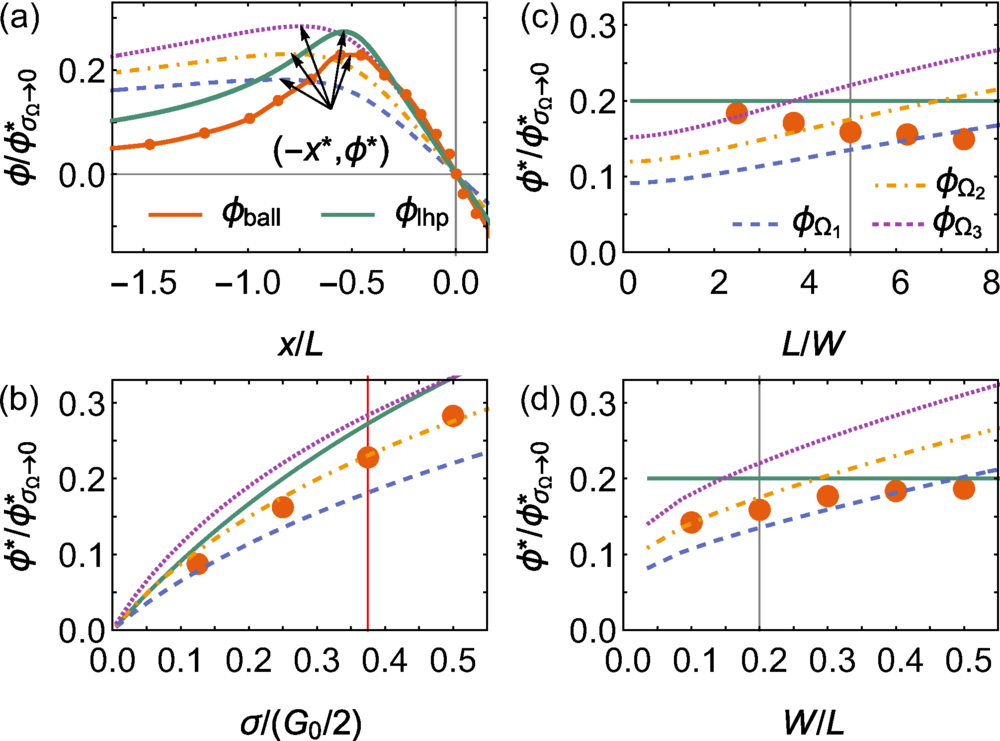
图 2:一维弹道边缘通道的归一化界面势,与几个电阻代理比较。
模拟显示,当电子在通道不同位置被注入时,电势梯度会出现弯曲或局部反转——这是理想 Landauer 模型下绝不可能发生的。
更贴合现实的验证
除开上述模拟,研究团队还提出可通过扫描隧道显微镜(STM)或纳米电压探针在实际实验中测得这种分布。这种电压曲线,就像电子留下的足迹,能区分理想弹道与部分耗散通道。
这一模型能解释许多实验中常见却无法用传统理论说明的现象——例如某些拓扑材料中,电流虽然保持高导通,但电压却表现出局部非线性。这不仅使理论更接近实验,也让研究者能区分真正的量子特性与实验伪影。
该模型表明,由于存在弹道通道,二维材料中的电流发生了根本性的变化。它预测了可以通过纳米探针或多尖端扫描隧道显微镜直接观察到的特征电压分布。