编辑丨%
在蛋白质组学分析中,已有不少大模型发挥着它们各自的能力。但基于质谱的大规模复杂数据集常会让桌面计算资源不堪重负,且需要手动配置分析。
FragPipe 是目前应用最广泛的蛋白质组分析平台之一,以速度快、定量准确著称,支持多种采集模式。但即使如此,它也在 HPC(高性能计算)或云环境中的实际部署面临着系统性障碍。
在这一背景下,来自匈牙利任自然科学研究中心(HUN-REN Research Centre for Natural Sciences)与美国纽约大学(NYU)等提出研究团队提出了 Frag’n’Flow ——一个专门为 FragPipe 设计的、面向大规模计算环境的自动化工作流。
相关研究内容以「Frag’n’Flow: automated workflow for large-scale quantitative proteomics in high performance computing environments」为题,于 2026 年 1 月 4 日发布在《BMC Bioinformatics》。

论文链接:https://link.springer.com/article/10.1186/s12859-025-06305-y
拆解化的工作流系统
在动辄几十几百 GB 的数据规模下,蛋白质组学分析所面临的问题往往不再是「能不能算出结果」,而是谁有能力把完整流程稳定地跑完。
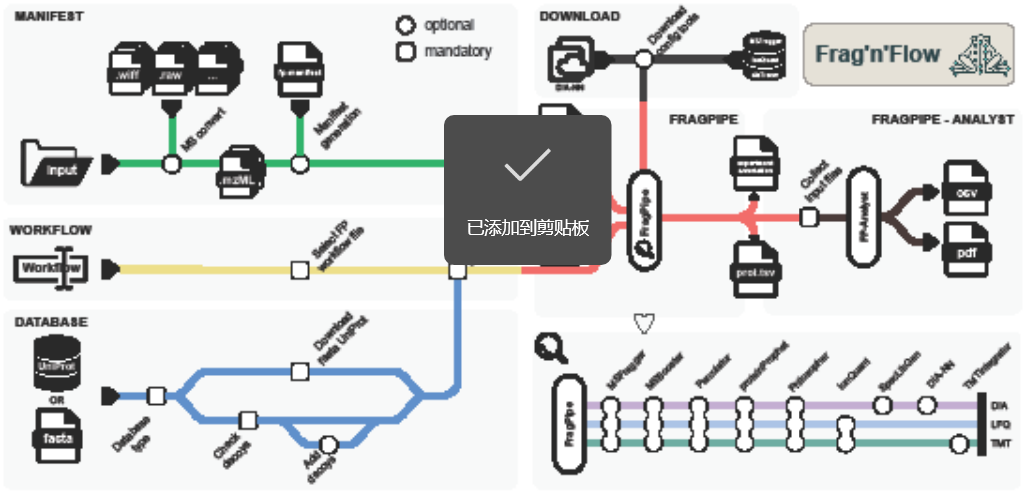
图 1:Frag’n’Flow 流程概述。
研究团队选择 Nextflow 作为工作流编排框架,将 FragPipe 及其关键组件(MSFragger、IonQuant、diaTracer 等)封装进一个可移植、可复现的流程中。
自动生成分析清单与流程配置,避免手工编写 manifest 文件;
通过容器化管理依赖,在 HPC、云平台和本地集群间保持一致行为,集成相关数据库;
支持 DDA、DIA 与 TMT 三类主流定量策略,选择合适的工作流;
配置工具下载模块,并内置下游统计分析模块,直接输出差异蛋白与通路结果;
在四个子模块就位后,、必要的输入文件、配置工具和环境设置会自动准备就绪,凭此无缝启动。因此,Frag’n’Flow 为高性能计算或云环境中部署提供了完全自动化的设置,最大限度地减少了人工干预,并确保了多次运行的一致性。
性能验证
实验验证部分专注于回答一个核心问题:在不牺牲定量准确性的前提下,Frag’n’Flow 是否真的提升了大规模分析效率?
因此,团队选择使用 quantms 来对 Frag’n’Flow的性能和输出结果进行基准测试。这两个工作流均使用公开的原始 DIA 数据集,采纳默认参数和标准的SLURM工作负载管理器。
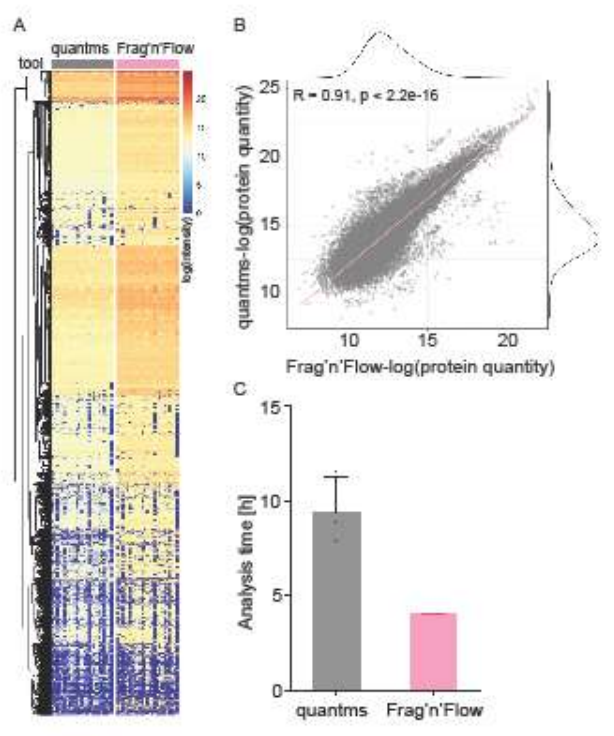
图 2:与竞争流程相比,Frag’n’Flow 具有高度一致性且运行时间更快。
该数据集大小大概在 58 GB,而处在这种数据规模下的 Frag’n’Flow 仍保持了定量准确性,且将运行时间几乎缩短了一半,同时缓解了内存和输入/输出瓶颈。团队在三个代表性数据集(无标记DDA、DIA和TMT)上验证了Frag’n’Flow 的结果,仅需极少的用户干预就能成功重现已发表的生物学特征。
通过整合 FragPipe-Analyst 的 R 实现版本,Frag’n’Flow 在主流程结束后自动完成数据归一化与缺失值处理;基于 limma 的差异表达分析;PCA、火山图、相关性热图等质量控制图;通路富集分析(Hallmark / KEGG)。这些结果以 CSV 与单一 PDF 报告形式输出,显著降低了分析门槛。
工程问题的系统解法
通过 Nextflow 的编排能力与 FragPipe 的高灵敏度结合,Frag’n’Flow 的研发团队将桌面导向的 FragPipe 转化为 HPC 适配的规模化工具,实现了从原始数据到生物学解释的端到端自动化分析。
该工作流程兼顾定量准确性与分析效率、支持多平台部署、遵循 FAIR 原则、操作门槛低,无需复杂手动配置。这是一种可扩展、可复现、对用户友好的实现路径,使大规模定量蛋白质组分析不再依赖复杂的人工配置。