作者 | 论文团队
编辑 | ScienceAI
2025 年 10 月,一篇题为《Hierarchical affinity landscape navigation through learning a shared pocket-ligand space》的论文,作为封面文章发表于 Cell 旗下期刊《Patterns》。该研究由粤港澳大湾区数字经济研究院(IDEA)AI4Science 团队、晶泰科技(XtalPi)及华盛顿大学的研究团队联合攻关,成功开发了名为 LigUnity 的亲和力基础模型。

论文地址:https://www.cell.com/patterns/fulltext/S2666-3899(25)00219-3?_returnURL=https%3A%2F%2Flinkinghub.elsevier.com%2Fretrieve%2Fpii%2FS2666389925002193%3Fshowall%3Dtrue
项目地址:https://idea-xl.github.io/LigUnity
开源代码:https://github.com/IDEA-XL/LigUnity
此项工作不仅是对传统虚拟筛选工具的优化,更是一次范式级的革新 —— 它通过构建蛋白质与配体的共享表示空间,让 AI 统一学习并驾驭从大规模筛选到精细活性优化的完整药物发现流程。
任务背景与挑战
在人体中,上万种蛋白质维持着复杂的生命活动,其中许多蛋白质的异常与疾病直接相关,是潜在的药物靶点。然而,目前仅有约 10% 的蛋白质能被已知的药物分子高效结合,绝大多数靶点仍处于「黑暗」 之中,等待着被有效的药物「点亮」。
为这些靶点找到合适的药物分子,通常分为两个关键步骤:
1. 虚拟筛选 (Virtual Screening):利用计算方法,从包含数亿甚至数十亿分子的巨大化合物库中,快速筛选出少数可能与目标蛋白质结合的「苗头化合物」。此阶段的核心诉求是速度。
2. 苗头化合物优化 (Hit-to-Lead Optimization):在找到苗头化合物后,需要对其化学结构进行精细的修改和优化以增强其活性(活性优化),最终得到可进入后续试验的「先导化合物」。此阶段的核心诉求是精度。
传统方法往往将这两个任务割裂开来,使用不同的工具和模型。这种分离导致了模型效果限制:专注于筛选的模型可能无法精确区分结构相似但活性差异巨大的分子;而专注于优化的模型则难以泛化到具有全新化学骨架的潜力分子。
模型方法与核心创新
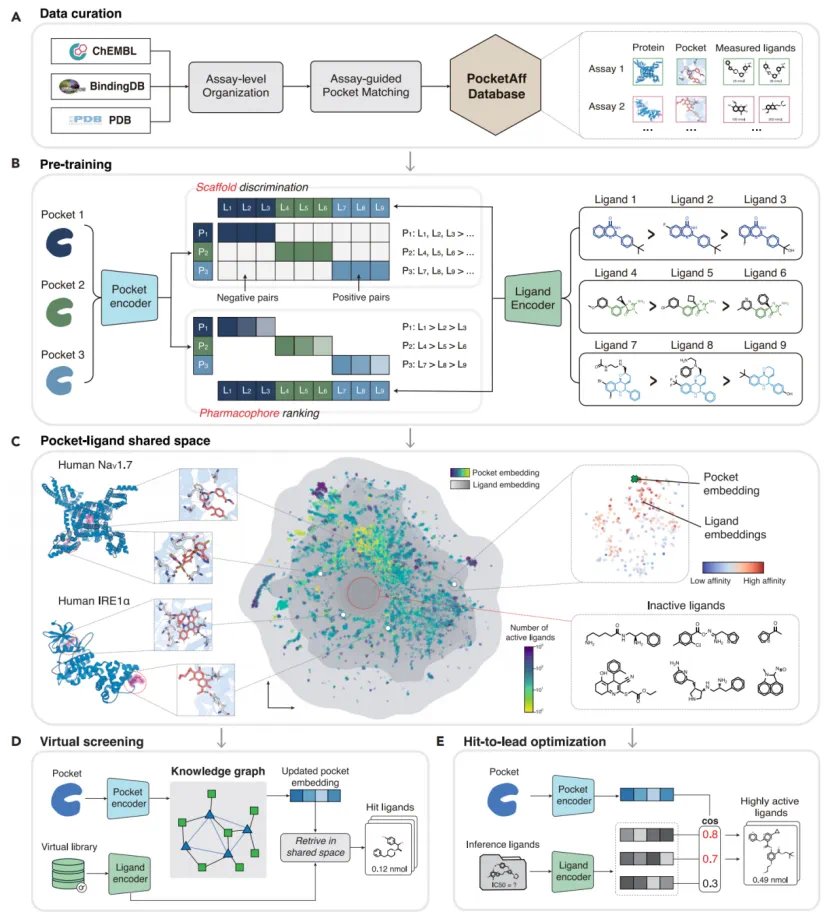
LigUnity 的核心创新在于构建了一个统一的亲和力基础模型,让 AI 学会一种能同时理解蛋白质结合口袋和药物分子的「共同语言」。
为实现这一目标,研究团队首先构建了迄今为止规模最大的、以实验(Assay)组织的亲和力数据库 PocketAffDB,其中包含了蛋白质口袋结构信息和近百万活性数据。基于此,LigUnity 通过一种创新的「层级式学习」策略进行训练:
1. 化学骨架判别:首先,模型利于对比学习(Contrastive learning)进行粗粒度的区分。它学习识别活性分子和非活性分子的骨架差异,为虚拟筛选任务打下基础。
2. 药效团排序:然后,模型利于列表排序(Listwise ranking)进行细粒度的优化。它学习对一群结构相似的活性分子,根据其活性的高低进行精确排序,以胜任苗头化合物优化任务。
在预训练期间,LigUnity 同时优化上述目标;在推理时,LigUnity 将蛋白和口袋映射至同一空间,使用余弦相似度计算其活性。在保证了高精度的同时,LigUnity 的速度比传统的分子对接方法(如 Glide-SP)快了百万倍。
实验结果
LigUnity 在横跨 6 大类应用场景的 8 个基准测试中,展现了其卓越的性能。
1. 虚拟筛选任务
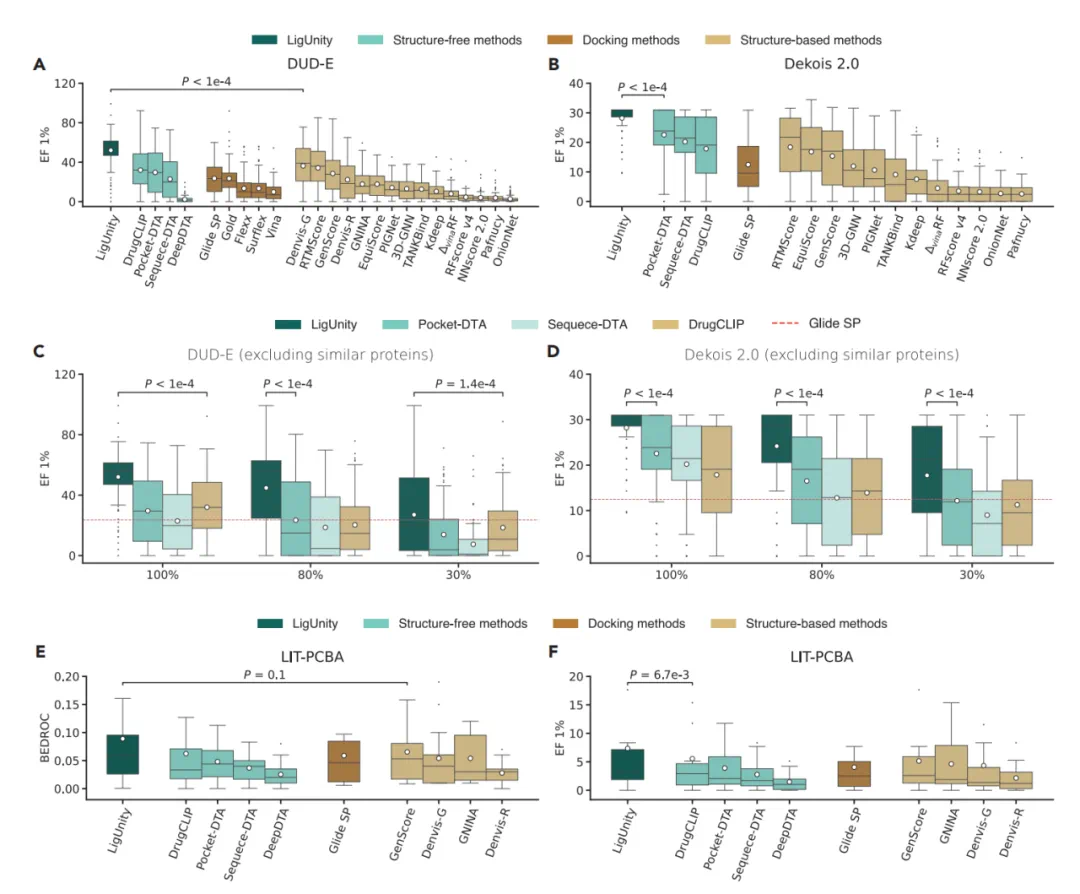
全面领先:在 DUD-E、DEKOIS 2.0 和 LIT-PCBA 这三个主流虚拟筛选基准测试中,LigUnity 的表现超越了全部 24 种竞争方法,包括传统的分子对接软件和各类机器学习模型。
性能巨大提升:与表现最好的基线模型相比,LigUnity 在关键指标「富集因子 (EF 1%)」上取得了超过 50% 的性能提升(p < 10⁻⁹)。
强大的泛化能力:即便在排除了与训练集中蛋白质相似(序列相似性 < 30%)的新靶点上进行测试,LigUnity 的性能依然显著优于 Glide SP,表现出对未知靶点的强大预测能力和实用价值。
2. 苗头化合物优化任务
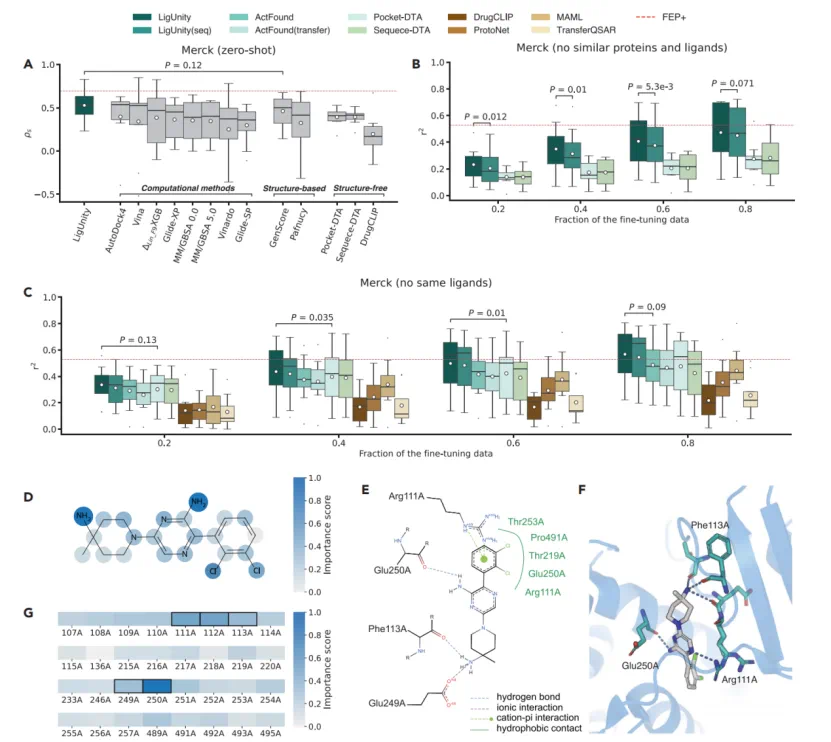
零样本性能优越:在 JACS 和 Merck 这两个行业公认的 FEP 基准测试中,在不使用任何目标数据(zero-shot)的情况下,LigUnity 的预测性能已优于 Glide-SP、MM/GBSA 等传统计算方法及其他机器学习模型,展现了其强大的预训练知识。
少量数据即可媲美「金标准」:当使用少量(10-20 个)已知数据进行微调后,LigUnity 的预测精度(在 Merck 基准上 r² = 0.472)已接近计算成本极度高昂的物理计算「金标准」方法 FEP+(r² = 0.528),为昂贵的实验和计算提供了高性价比的替代方案。
良好的可解释性:尽管未使用蛋白 - 分子结合构象作为输入,LigUnity 仍成功识别出了对结合起关键作用的配体原子和口袋残基,与已知的晶体结构相互作用模式高度吻合,为化学家优化分子提供了可靠指导。
3. 多场景应用的通用性
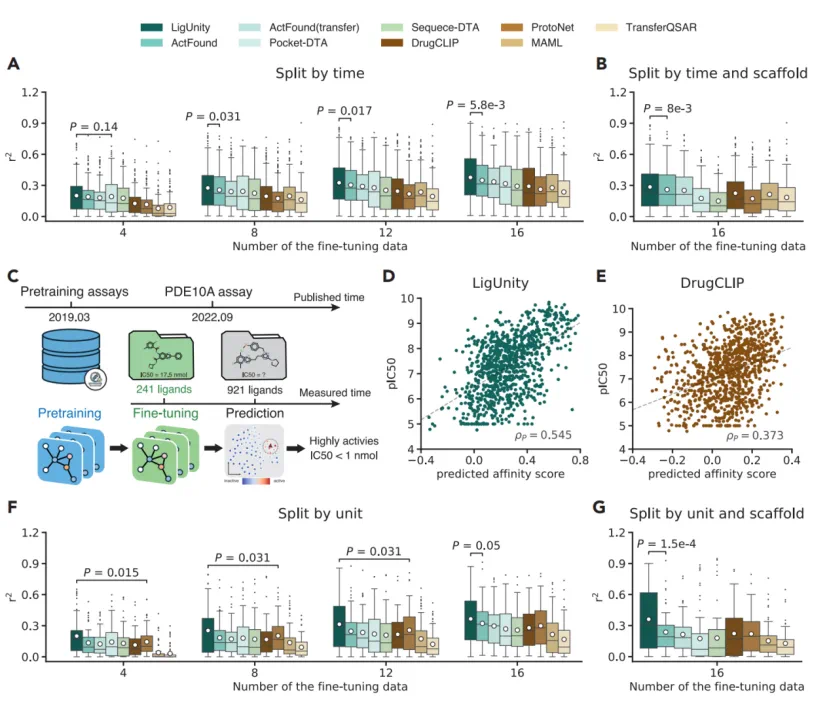
前瞻研究与骨架跃迁:在模拟真实药物发现的测试中,LigUnity 表现出众。在时间分割(用历史数据预测未来新分子)和化学骨架分割(泛化至训练集中未见的全新化学骨架)设定下,它均超越了所有基线模型,证明其具备突破现有化学空间、发现新潜力分子的强大泛化能力。
对多源、异构数据的鲁棒性:真实世界的生物活性数据来源多样、测量单位不一(如 nM vs. % 抑制率)。得益于其独特的「列表排序」机制,LigUnity 天然的对测量单位不敏感。在 OOD 测试中,其性能相比传统回归模型提升高达 40.2%,展现了其作为基础模型处理复杂、真实世界数据的卓越适应性。
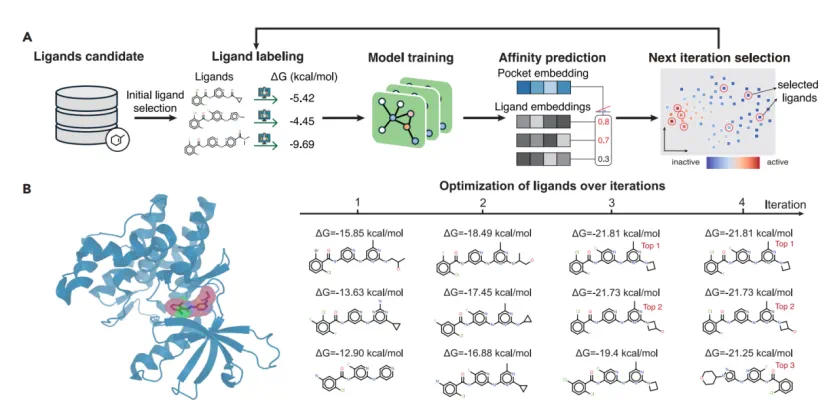
主动学习应用:在模拟真实研发流程的主动学习框架中,研究团队对 TYK2 靶点进行研究,从 10,000 个后续分子中找到活性最好的分子。集成 LigUnity 的框架仅通过 4 轮迭代(400 活性标注数据),就成功找到了活性最高的 3 个分子,证明其能以极少的实验成本,高效地指导药物发现进程。
结论与展望
LigUnity 通过一个统一的、层级式的学习框架,成功地将虚拟筛选与活性优化相结合,在速度、精度和泛化性上均取得了突破。它不仅是一个强大的预测工具,更是一个具备广泛适用性的基础模型,为加速早期药物发现的全流程奠定了坚实的基础。