编辑丨&
现实里,分子是不断运动的,它们的相遇与结合像是一部动态电影,而这也就为药物研发带来了艰巨的挑战:现有方法对这种复杂运动视而不见,模型在“标准基准”上看似成绩斐然,却往往在真实药物场景下失灵。
亲和力预测模型大多依赖公开数据集 PDBbind,其中约 2 万个复合物结构成了 AI 的训练教材。但这类训练存在明显「信息泄漏」:模型可能学会的是数据集的特征,而非真实的物理规律。
为此,来自法国奥尔良大学(Université d’Orléans)的研究团队引入了分子动力学模拟(MD)。这种方法能在原子层面追踪蛋白与配体的运动,获得数十纳秒甚至更长时间的轨迹。
他们的研究以「Spatio-temporal learning from molecular dynamics simulations for protein-ligand binding affinity prediction」为题,于 2025 年 8 月 19 日发布在《Bioinformatics》。

论文链接:https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btaf429/8238154
两条轨迹齐头并进
准确预测蛋白质-配体复合物的结合亲和力是药物设计的一个主要目标。但从目前发布的模型来看,它们对性能的预评估都相当乐观,应用于新的或者更复杂的测试集的时候,难免会因为泛化性较低导致限制使用。
所以,研究团队就提出了两种融合 MD 与深度学习的策略。
第一条是数据增强路线:从分子动力学轨迹中抽取帧,扩充训练数据。研究者构建了一个全新的数据集——MDbind,涵盖 6300 个蛋白-配体复合物,每个跑 10 次 10 ns 模拟,总计 63,000 段轨迹。从这些轨迹里抽帧,就能把原本不足 2 万的样本扩展到数百万。
该方案的目标之一是使 DL 模型能够区分高亲和力和低亲和力配体之间的差异,并捕获它们相互作用的变化,而大多数在静态单一 3D 蛋白质-配体复合物上训练的模型常常无法捕捉到这些。
图 1:分子动力学模拟。
第二条是时空学习路线:让模型直接学习整段轨迹。研究团队设计了 Timenucy(基于长期递归神经网络 LRCN) 与 Videonucy(基于卷积长短期记忆网络 ConvLSTM),这类架构能同时捕捉空间结构和时间演化,分析模拟的所有帧以进行结合亲和力的预测。
这两者将会在整个模拟中进行训练,从使用3D输入数据过渡到使用4D输入数据。两者都处理每个原子的信息,包括它们的特征和位置,针对模拟的每一帧。
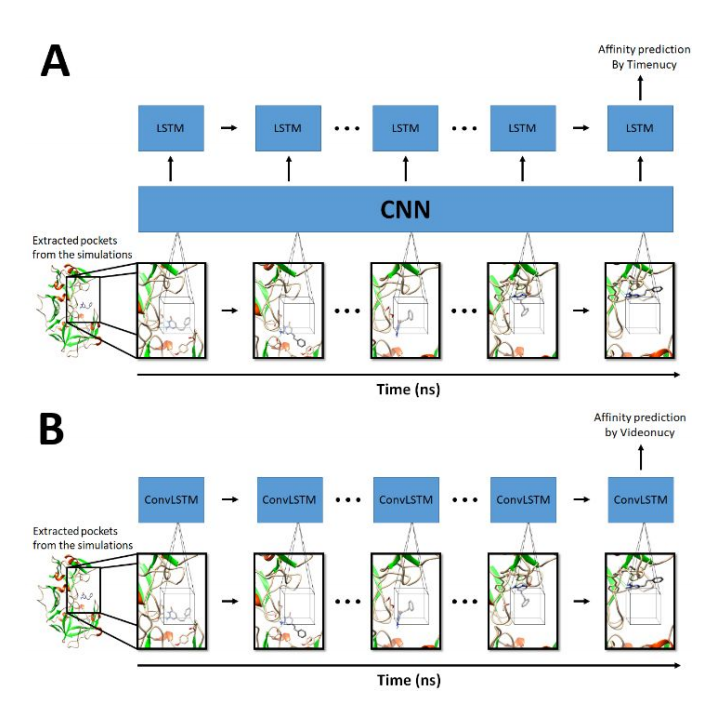
图 2:Timenucy (A) 和 Videonucy (B) 的工作流程。
用数据说话
在经典基准 PDBbind v2016 core 集合上,加入 MD 增广的 Densenucy 模型取得了 R≈0.83,RMSE≈1.28 的表现。这一成绩不仅超越了无增广的版本,也明显优于历史上的其他模型。
更令人兴奋的是在更接近真实药物研发的外部测试集上——尤其是包含“陡峭活性崖”的 FEP 数据集。在这类场景中,同一靶点的分子往往因为微小结构差异,亲和力就会骤升或骤降,考验模型的泛化能力。
p38 激酶簇上,传统模型 KDEEP 的表现是 R=0.36、RMSE=1.57;而 Densenucy+MD 增广提升到 R=0.66、RMSE=0.62。在多个其他簇中,带 MD 增广的模型普遍表现更稳健。
表 1:模型在 PDBbind v.2016 核心集和 MDbind 测试集上的性能。
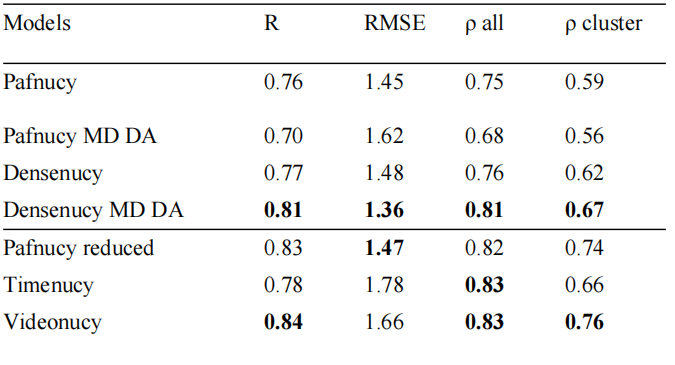
研究团队表示,由于在 PDBbind 上训练并在 PDBbind v.2016 核心集上评估的模型性能存在偏差,他们决定使用外部测试集 FEP 数据集进一步对模型进行基准测试。Timenucy / Videonucy 这类时空模型在 MDbind 上也能学习到差异,虽然目前性能尚未超过最优静态模型,但已经证明了这条路线的可行性。
小结
如果说传统的 AI 药物模型只是在静态照片上做“图像识别”,那么这项研究展示了另一种可能:让模型真正看懂分子的活动轨迹。63,000 段模拟不仅刷新了训练规模,也让模型更接近真实的物理过程。
这就像是在培养一个人造的拉普拉斯妖——读懂每一个分子,接着它就可以预测未来。
听着很魔幻,但科学就是如此,将理论转变为现实。正如研究团队所强调的,这只是第一步。未来,随着更高效的时空模型与更丰富的轨迹数据出现,AI 将能在更大范围内把握分子间的复杂互动,帮助科研人员在药物研发这条路上少走弯路。